



引 言
中国是一个富煤、少油、缺气的国家,是世界上最大的煤炭生产和消费国,煤炭在我国国民经济和社会发展中占据重要地位。在我国碳中和历程中,煤炭是不可或缺的板块[1]。如何高效洁净地利用煤炭资源,合成燃料及高品质化学品是我国能源领域的重大课题。
迄今为止的煤化工基本上都属于热化学加工,煤热解是煤转化过程的第一步,对煤的后续转化(气化、液化、燃烧和炭化)有重要影响[2-3]。煤的热解是煤在隔绝空气或惰性气氛的条件下被加热至一定温度而发生的物理变化和化学反应的复杂过程[4]。认识煤热解的初始反应机理是对煤热解和其他转化过程进行调控和清洁利用的基础和关键。但目前人们对煤快速分解反应的机理认识仍停留在概念表述和经验范畴[5]。一般认为,煤热解反应机理可分为两个步骤[3, 6]:(1)煤中部分弱共价键(主要为脂肪桥键和醚键等)受热分解产生自由基碎片,包括挥发分自由基和游离的小分子自由基(•OH, •H, •CH3等);(2)部分挥发分自由基碎片若能从煤基网络孔道中逃逸出去,会被小分子自由基稳定化生成挥发分产物(stabilization of radicals),若挥发分自由基的尺寸较大,无法迁移至孔道表面,则会彼此连接发生缩聚或交联反应生成固体产物(polymerization and condensation reaction)。
为此,挥发分自由基涉及的被稳定反应和缩聚反应不仅影响焦油产物的性质和收率,也对焦油后续进一步加工提质生产液体燃料或高品质化学品至关重要。刘振宇团队[7-10]针对挥发分自由基行为开展了一系列研究,探索了自由基浓度、挥发分二次反应及其对气体生成的影响。其中Zhou等[10]利用自旋(顺磁)共振仪(electron spin resonance,ESR),通过测定稳定自由基浓度发现低阶煤和次烟煤在440~700℃温度范围内挥发分涉及的反应并不完全相同,所获得的稳定自由基浓度随温度上升的现象则说明高温促进缩聚反应,使得固体产物芳香度增加、含氧减少。He等[7]借助ESR研究了四种煤样在不同溶剂(环己烷、四氢呋喃)和不同温度下(300、350、400和450℃)的热解过程中焦油的自由基浓度,结果表明煤阶越高,焦油中挥发分自由基浓度越高。尽管目前有很多工作聚焦于煤热解的工业生产过程,但前人对煤结构和反应的分子水平认识仍停留在猜测层面。只有在研究思路上另辟蹊径,才有可能找到突破口,推进煤热解反应机理认识的发展。
煤的分子结构和煤热解过程均非常复杂[13-14]。尽管ESR目前可检测煤热解过程中被固体产物包围的自由基浓度,但检测的时间尺度远远大于挥发分活性自由基的存活时间,且无法确定自由基的种类及其随时间的动态演化,更无法直接获得挥发分自由基在煤热解过程涉及的化学反应,区分挥发分自由基涉及的被稳定反应和缩聚反应则更加困难。随着计算机能力的持续提升,能够模拟微观反应过程的反应分子动力学方法(reactive molecular dynamics, ReaxFF MD)正成为认识煤热解挥发分自由基生成与反应机理的重要途径。反应分子动力学是将化学反应力场(reactive force field)与分子动力学(molecular dynamics, MD)相结合的方法,其中由van Duin等[15-16]提出的反应力场ReaxFF有潜力应用于大规模煤模型的热解模拟。ReaxFF是一种基于键级的经验力场,原子之间的成键及氢键相互作用均为键级的函数,可连续描述键的生成和断裂;通过在每个时间步对原子的部分电荷进行动态优化,较好地考虑了极化作用;并利用量子化学计算和实验数据的拟合,将原子和原子之间的相互作用参数加以优化,得到了一个较其他基于键级方法更为准确、具有反应性的经验反应力场。与量子化学中应用广泛的密度泛函方法(density functional theory, DFT)相比,ReaxFF MD将可模拟的体系规模提高至少一个数量级,且其计算精度与DFT较为接近。由于煤分子结构复杂,其热解过程多路径反应同时进行,很难人为预先假设反应路径,ReaxFF MD恰好具有无须预设反应路径、模拟反应过程的优势,并且已具备描述煤结构中主要元素C、H、O、N、S的统一力场[17],使其成为研究煤热解挥发分自由基及其化学反应机理的新途径。
目前,ReaxFF MD方法已被广泛应用于复杂的煤热解和燃烧过程[18-22]。van Duin等[19, 23]利用ReaxFF MD在煤热解机理研究上进行了最早尝试。Castro-Marcano等[17,24]模拟了含7458个原子的伊利诺伊6号煤焦模型的燃烧,及含50789个原子伊利诺伊6号煤模型在2000 K的热解;模拟探索了硫元素对煤热解的影响,讨论了不同类型的含硫官能团在热解过程中各自发挥的作用,说明该力场具有研究大规模煤模型高温热转化过程的潜力。本文作者所在团队也开展了与煤热解相关的ReaxFF MD模拟探索,并发展了ReaxFF MD模拟大规模煤热解的新方法。为了在可接受的计算时间内模拟超过10000个原子的大规模煤模型,创建了基于图形处理器GPU并行的反应分子动力学程序软件GMD-Reax[25],直接模拟了含有98748个原子的淖毛湖煤模型[26]在500~2500 K的热解过程;利用用于ReaxFF MD模拟结果化学反应及可视化程序系统VARxMD (visualization and analysis of reactive molecular dynamics)[27]获得了煤热解产物全景式演化规律和与之对应的反应机理。
更为重要的是,作者团队在ReaxFF MD应用于煤高温热转化的合理性和可信度验证上进行了大量的工作。通过对比柳林烟煤的快速热解实验(Py-GC/MS)在673~1073 K的热解主要产物和含有28351个原子柳林烟煤模型在1000~2600 K的ReaxFF MD模拟结果,表明ReaxFF MD模拟的柳林烟煤热解过程可获得与热解实验一致的萘及其衍生物的生成趋势[28]。通过对含有23898个原子、多组分府谷次烟煤模型的ReaxFF MD热解模拟与热解同步辐射真空紫外光电离飞行时间质谱(pyrolysis synchrotron radiation vacuum ultraviolet photoionization time-of-flight mass spectrometry, Py/SVUV-PI-TOF-MS)实验获得的主要热解产物及其演化规律进行比较,发现基于大规模多组分煤分子模型的ReaxFF MD模拟煤热解过程可以获得与Py/SVUV-PI-TOF-MS实验基本相同的热解产物组成,和高度相似的代表性热解产物C2H4的时间演化趋势[29-30]。同时,通过对海拉尔褐煤在Py/SVUV-PI-TOF-MS、Py-GC/MS热解实验和ReaxFF MD模拟结果的对比,表明模拟不仅可以获得与实验一致的产物分布和演化规律,还可以揭示产物的生成消耗反应机理和反应网络[31]。这些与实验对照的模拟结果均验证了ReaxFF MD模拟大规模煤热解模型的合理性。
基于介尺度理论和大规模ReaxFF MD模拟[32]的优势,本文结合基于GPU的高性能计算和化学信息学分析反应机理的方法,深入研究了煤热解挥发分自由基的作用,在系统考察微观尺度煤热解过程中的本征化学反应和产物的相互对应关系的基础上,聚焦挥发分自由基在逸出过程中被稳定反应和缩聚反应及这两类化学反应随时间、温度的动态演化规律和竞争协调机制,以期揭示煤热解反应控制机制可预测焦油和焦炭收率的特性,为碳中和社会下煤化工的高效转化和清洁利用提供理论支持。
1 模拟方法
1.1 模型构建和ReaxFF MD模拟细节
表1 ReaxFF MD模拟策略和细节
Table 1
| 序号 | 策略 | 模拟温度 | 模拟时间 | 目的 |
|---|---|---|---|---|
| 1 | 慢速升温 | 500~2500 K | 1 ns | 挥发分自由基的阶段性特征 |
| 2 | 直接恒温 | 1600、1900、2000、2200 K | 1 ns | 温度对挥发分自由基行为的影响 |
| 3 | 先升温后恒温 | 1800、2000、2200 K | 恒温400 ps | 减少弱键断裂的影响,挥发分自由基在不同反应阶段后期的行为 |
首先,采用2 K/ps的速率实现大规模柳林烟煤的升温模拟,模拟温度为500~2500 K,时间为1 ns。基于在木质素和煤热解的模拟结果发现[26, 34-36],2 K/ps对于模拟而言是较慢的升温速率,可全景式获得反应物和产物在不同热解阶段的行为和分布,进一步揭示挥发分自由基的阶段性特征。其次,为了研究温度对挥发分自由基的影响,对大规模柳林烟煤模型开展了1600、1900、2000和2200 K的ReaxFF MD恒温模拟,模拟的持续时间为1 ns。此外,通过2 K/ps升温ReaxFF MD模拟发现,1400 K是柳林烟煤弱键断裂和一次裂解的起始温度,挥发分自由基从该温度开始生成。为了尽可能减少煤结构中弱键断裂对挥发分自由基行为的干扰,考察挥发分自由基在不同阶段反应后期的行为,本文增加了表1中序号3的三组模拟,即分别从500 K升温至恒温模拟的目标温度1800、2000和2200 K,然后再进行相应温度的恒温模拟,并重点考察恒温模拟阶段的化学反应,恒温阶段的反应停留时间为400 ps。其中,1800、2000和2200 K分别对应柳林烟煤一次裂解和二次裂解的转折温度、主要二次裂解阶段和聚合结焦为主阶段的起始温度,由此获得的挥发分自由基更具代表性。
所有的ReaxFF MD模拟均采用周期性边界条件和NVT系综,选用Berendsen控温方法,温度阻尼常数为0.1 ps;模拟采用velocity-Verlet算法动态更新原子在每个时间步的坐标和速度,时间步长为0.25 fs。所有的ReaxFF MD模拟均采用Mattsson反应力场。其中,Mattsson力场已被证实可合理描述煤、木质素、纤维素的热解过程[28, 35-37],获得的主要热解产物(焦炭、焦油和气体)和特定产物(小分子气体、苯、苯酚、萘等)演化趋势与大量实验获得的结果一致。值得注意的是,所模拟的反应时间尺度(ps级)比实验时间尺度(s级)相差多个数量级,因此模拟温度采用了“提温”策略,即模拟温度被人为地提升到一个比实验温度(500~1000 K)高出很多的温度,使得大部分反应都能够在可接受的模拟时间内发生。虽然ReaxFF MD模拟的时间尺度和温度都与实验有所不同,“提温”仍然是ReaxFF MD广泛采用的模拟策略,已有研究[26, 38]从ReaxFF MD模拟中得到了很有价值的结果,包括可与实验进行比较的反应产物,以及实验无法得到的自由基反应机理。
1.2 反应分析策略
VARxMD是本文作者课题组自主研发的ReaxFF MD反应分析与可视化软件,其独特之处是通过原子键类型识别实现了反应位点的识别,反应分析的所有结果均建立在反应位点识别之上,具有化学反应列表的生成、反应体系3D可视化显示、基于物种和化学反应位点结构等特征检索产物和反应类型、从某一指定反应物到指定生成物之间复杂反应网络的自动遍历生成等独特功能[39-40]。VARxMD近期功能[41]进一步扩展了可自动追踪指定局部反应区域内的结构演化、化学反应和中间体,分析体系结构特征动态信息的功能。借助VARxMD分析可知,对在约100000个原子规模的柳林烟煤模型进行1 ns高温恒温模拟过程中,共发现超过200000个反应发生,涉及超过100000个物种。从如此庞大的化学反应数据中找到挥发分自由基及其参与的化学反应十分具有挑战性,为此本文采用了分层次分析的检索策略。
本文将C5~C40的不饱和物质视为挥发分自由基。为了获得挥发分自由基参与的化学反应随时间温度的演化趋势,首先利用VARxMD比较产物的不饱和度,从模拟体系的100000个物种中检索出所有的挥发分自由基;其次以挥发分自由基作为反应物,从200000+个化学反应中找到所有以“挥发分自由基作为反应物”的化学反应,得到挥发物自由基参与的反应,作为进一步追踪生成挥发物自由基反应去向的子反应列表。从煤热解的反应性质可知,挥发分自由基被稳定化的反应会消耗含氢小分子自由基,例如•OH、•CH3,并进一步生成焦油产物(C5~C40);与此同时,挥发分自由基的缩聚反应会生成大片段物质,即焦炭和碳烟(C40+物质)。为此,基于“挥发分自由基作为反应物”的子反应列表,通过检索产物为C40+物质并且反应位点发生C—C键/ C—O键生成的化学反应,得到挥发分自由基参与的缩聚反应;通过检索反应物为C0~C1含氢小分子自由基且产物为C5~C40物质的化学反应,得到挥发分自由基参与的被稳定反应。本文采用的分层检索策略基于VARxMD独特的可对化学反应按物种和反应位点分类的功能,所获得的煤热解反应机理认识是其他基于量子化学计算方法和其他ReaxFF MD反应分析平台无法获得的。
2 结果与讨论
2.1 煤热解慢速升温过程中反应的竞争与协调
柳林烟煤模型2 K/ps升温的ReaxFF MD模拟(500~2500 K)结果表明:柳林煤主要裂解阶段从1400 K开始,聚合结焦为主的阶段从2400 K开始。图1给出了分层次分析策略获得的柳林烟煤模型在主要热解阶段1400~2400 K温度范围内挥发分自由基参与的稳定化为焦油和发生缩聚或交联这两类化学反应的数量的演化趋势,同时给出了与此密切相关的煤的失重和焦油收率在该温度范围内随温度的演化规律。其中,圆形标志表示挥发分自由基参与的被稳定反应和对应的焦油收率,方框标志代表其缩聚反应和大片段物质的质量分数演化。如图1(a)所示,挥发分自由基参与的生成焦油和缩聚为大片段的两类化学反应在整个烟煤主要热解过程中竞争激烈。在煤热裂解的一次反应初期(1400~1600 K),缩聚反应数量略多于被稳定反应。这是由于煤裂解初期生成挥发分自由基还没来得及从煤基孔道中逸出,更容易彼此连接、发生缩聚反应;游离在煤基孔道之外的小分子自由基能够捕捉挥发物自由基的机会不多。随着温度升高,获得能量的挥发分自由基更容易从煤基网络逃逸出来,有更多机会被含氢小分子自由基捕捉,从而被稳定反应的数量超过了缩聚反应。图1(a)表明挥发分自由基被稳定反应的数量与缩聚反应的差距在1550~1800 K随着温度上升,在1800 K到达差距最大值之后在1800~2200 K随温度下降。值得注意的是,挥发分自由基从被稳定化占主导转向缩聚占主导所对应的1800 K这一转折温度恰好与柳林烟煤热解的一次裂解阶段向二次裂解阶段的转折温度相一致,表明挥发分自由基及其参与的这两类化学反应可代表或决定煤热解的阶段性特征。当温度继续升高至2200~2400 K,被稳定反应和缩聚反应竞争非常激烈,无法判别出挥发分自由基涉及的哪类反应在煤热解过程中占主导,但缩聚反应数量从2350 K开始有超过被稳定反应的趋势。
图1

图1
由ReaxFF MD和VARxMD获得的煤热解升温过程中挥发分自由基参与的被稳定和缩聚反应随温度的竞争协调趋势及与此相关的焦油/焦炭产物的质量分数演化(图1(b)是基于文献[22]中数据的重新绘制,Copyright 2017, Taylor & Francis)
Fig.1
Evolving trends of reactions with volatile radicals involved and the mass percentages of the related char/tar products obtained from ReaxFF MD and VARxMD (Fig.1 (b) was replotted based on the data of Ref. [22]. Copyright 2017, Taylor & Francis)
由图1(b)可知:挥发分自由基参与的被稳定化和缩聚两种控制机制与煤热解主要产物焦油/焦炭的收率密切相关,在柳林烟煤快速失重且挥发分自由基被稳定反应占主导的阶段(1500~2200 K),焦油产物(C5~C40)大量生成;而当温度升高至这两类化学反应类型竞争激烈并出现缩聚反应占主导的趋势时(2200~2400 K),焦油的生成速率开始减慢并在2350 K出现从增长到不再增长对应平台的转折,即焦油收率到达其最大点。因此,挥发分自由基涉及的化学反应类型并不单一决定产物的组成和收率,其两个反应控制机制的竞争和协调共同决定了产物分布,特别是焦油和焦炭产物的组成和收率。
2.2 恒温过程中煤热解反应的竞争与协调
由于2 K/ps升温模拟会使得反应物和中间体在每个温度停留时间过短,反应没能完全发生。为此,本节基于柳林烟煤开展了长时间恒温模拟,模拟温度分别为1600、1900、2000和2200 K,模拟时间为1 ns。由2.1节的升温模拟结果可知,挥发分自由基参与的被稳定反应和缩聚反应是控制煤热解三阶段的特征反应,借助VARxMD可对反应进行自动分类和分层次检索策略,将恒温模拟得到的挥发分自由基参与的被稳定反应和缩聚反应进行计量,进而获得这两类反应数量在不同温度下随时间的演化规律,如图2所示。相应地,与这两类反应密切相关的焦炭(C40+)和焦油(C5~C40)产物质量分数在不同温度下随时间的变化趋势如图3所示。
图2

图2
由长时间ReaxFF MD恒温模拟获得的在柳林烟煤热解过程中挥发分自由基参与的被稳定反应和缩聚反应随时间的竞争演化规律
Fig.2
Number evolving tendencies of condensation reactions and stabilization reactions with volatile radicals involved obtained from long-time iso-thermal ReaxFF MD simulations at different temperatures
图3
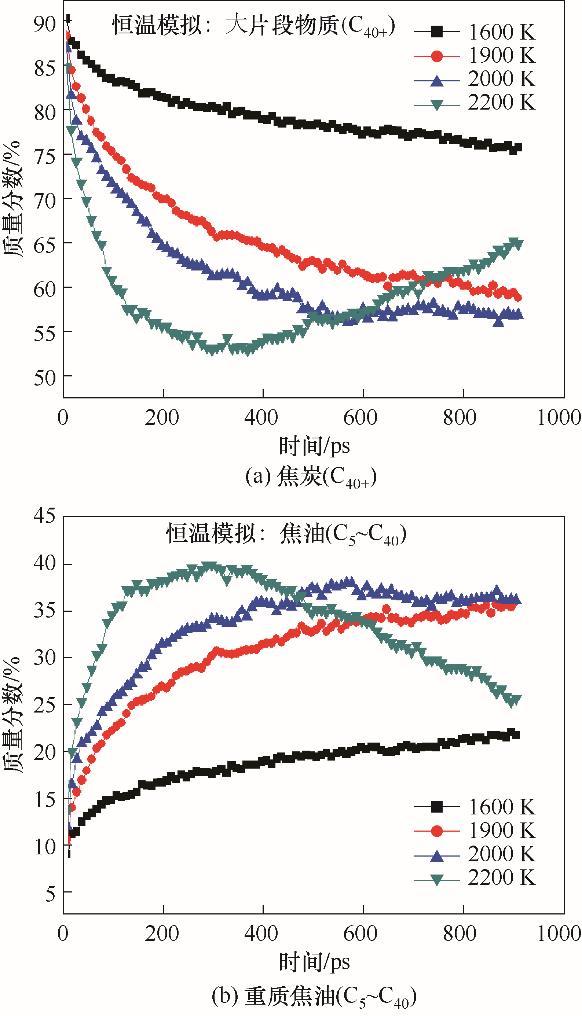
图3
由ReaxFF MD恒温模拟获得的柳林烟煤热解过程中主要产物质量分数在不同温度下随时间的演化规律
Fig.3
Evolving tendencies of mass percentages for major pyrolyzates with time obtained from long-time iso-thermal ReaxFF MD simulations at different temperatures
如图2(a)所示,在1600 K的较低温阶段,挥发分自由基参与的被稳定反应的数量始终比缩聚反应多5~10个,在整个模拟过程占主导,使得图3所示的C40+质量分数随时间持续下降,焦油收率持续上升。由此可知,虽然两类化学反应竞争激烈,但在温度较低的裂解阶段(温度低于升温模拟中一次裂解阶段和二次裂解阶段的转折温度1800 K时),挥发分自由基被稳定反应始终占主导地位。当温度升高至1900 K时,挥发分自由基参与的被稳定反应在0~500 ps占主导,但其数量随时间逐步降低,与缩聚反应数量的差距在缩小。如图2(b)所示,在模拟的500~1000 ps阶段,挥发分自由基涉及两类反应数量接近,无法区分谁占主导,由此带来了图3(b)展示的焦油质量分数在1900 K、500~1000 ps增长速度十分缓慢。值得注意的是,在1900 K的模拟末期(800~1000 ps),挥发分自由基涉及的缩聚反应数量逐渐增多,并有多于被稳定反应的趋势。
如图2(c)所示,挥发分自由基参与的反应控制机制随时间的演化具有明显的三区域特征:即在2000 K的前100 ps被稳定反应略多于缩聚反应;在100~500 ps两类反应控制机制竞争激烈;从500 ps开始发生主导反应(控制机制)的转变,即缩聚反应开始占据主导,缩聚反应的数量开始多于被稳定反应。该现象表明挥发分自由基在高温或者反应后期更容易彼此连接,使得焦油收率在2000 K的反应后期(500~1000 ps)略微下降,焦炭质量分数略微上升,如图3所示。在模拟温度为2200 K时,柳林烟煤热解呈现出聚合结焦为主的阶段[图2(d)],挥发分自由基参与的缩聚反应数量远多于被稳定反应,占绝对主导,很大程度决定了焦炭和焦油在高温的收率演化。在高温的整个模拟阶段,柳林烟煤结焦剧烈,焦炭质量分数从该温度的300 ps左右便开始显著上升,焦油含量相应地呈现快速降低的趋势。
恒温模拟结果一方面支持了升温模拟揭示的挥发分自由基参与的被稳定和缩聚两类主导化学反应,同时也表明这两类化学反应在煤热解不同阶段具有不同的竞争关系和主导作用。被稳定反应占主导的区域发生在烟煤热解的较低温阶段,使得焦油收率升高,但尚未达到收率最高值;相应地,缩聚反应占主导的区域出现在高温阶段,使得煤热解的结焦剧烈;当煤热解处于二次裂解主导阶段时,被稳定反应和缩聚反应竞争激烈。特别重要的是,当挥发分自由基参与的化学反应演化关系处于转折阶段时更容易出现焦油和焦炭收率的转折点,即被稳定反应占主导转折至被稳定和缩聚反应竞争激烈可对应焦油收率的最高值,两类反应竞争激烈转折至缩聚反应占主导可出现焦炭生成的起始点。该现象说明挥发分自由基主导反应的演化控制着煤热解反应的竞争与协调特性,可作为焦油和焦炭产物分布预测的重要标志。
2.3 挥发分焦油在二次反应过程中的竞争与协调
升温和恒温模拟结果表明,柳林烟煤热解过程具有明显的阶段性特征,其中由2 K/ps升温模拟获得的一次裂解向二次裂解阶段的转折温度1800 K对认识挥发分自由基的反应行为非常重要。为了深入探索挥发分自由基对焦油产物生成的影响,采用了升温无缝连接恒温模拟的连续模拟策略考察柳林烟煤的热解特性,即从ReaxFF MD升温至主要热解阶段的1800、2000和2200 K,分别开展了1800、2000和2200 K的400 ps恒温模拟。所选取的三个温度为一次裂解向二次裂解的转折温度或高于转折温度,均高于柳林烟煤模型在升温模拟中的一次裂解起始温度(1400 K),这意味着在恒温模拟之初体系中即存在大量挥发分自由基,该模拟策略可尽可能减少煤结构弱键断裂反应对挥发分自由基反应行为研究的干扰。图4给出了挥发分自由基参与的被稳定反应和缩聚反应各自所占比重,及焦油的质量分数在三个温度下随时间的演化规律。
图4

图4
由ReaxFF MD模拟获得的挥发分自由基在不同反应阶段后期的两类反应控制机制(被稳定反应和缩聚反应)在不同温度下恒温阶段随时间的演化规律
Fig.4
Number evolving tendencies of condensation reactions and stabilization reactions with volatile radicals involved at late pyrolysis stage obtained from ReaxFF MD simulations at different temperatures
如图4(a)所示,挥发分自由基在1800 K的整个模拟过程都更容易被含氢小分子自由基捕获,发生被稳定反应,使得焦油收率随时间一直上升。值得注意的是,虽然被稳定反应数量在整个热解阶段均比缩聚反应数量多,但随时间略微下降,与缩聚反应数量差距缩小。可以推测,若延长模拟时间,缩聚反应所占比重可能会超过50%,发生主导反应机制的翻转。如图4(b)所示,当温度升高至2000 K,挥发分自由基参与的被稳定反应和缩聚反应数量非常接近,并明显存在三个竞争区域,即被稳定反应占主导区域、两类反应竞争区域和缩聚反应占主导区域。挥发分自由基在2000 K的前150 ps更容易被小分子自由基稳定,使得焦油收率增长。在反应中段150~300 ps,两类反应控制机制的比重几乎完全相同,在竞争中协调,无法区分主导反应类型;在该区域,焦油收率从150 ps不再增长,证实了获得的结论,并说明被稳定和缩聚两类化学反应竞争协调的区域与焦油产物的最大收率非常一致。随后,缩聚反应在300~400 ps略微占主导。
如图4(c)所示,挥发分自由基的缩聚反应在2200 K的高温占绝对主导,其数量随时间振荡增加,而被稳定反应的数量随时间持续减少。挥发分自由基参与的缩聚反应使得重质焦油在100~400 ps持续下降,说明了比一次裂解阶段到二次裂解阶段转折温度高很多的温度并不适合获得高收率的焦油产物。综上可知,焦油的最高收率存在于被稳定反应占主导转折至被稳定和缩聚反应竞争激烈的区域或温度范围。若可延长挥发分自由基被稳定反应的阶段,或抑制其参与缩聚反应的阶段,对工业生产获得高收率焦油,以生产更多高品质化学品很有帮助。
3 结 论
挥发分自由基在煤热解过程中发挥着重要作用。基于介科学理论中复杂系统存在“不同主导机制之间在竞争中的协调”的共性规律,本工作聚焦煤热解过程中挥发分自由基(基本单元)及焦油/焦炭分子群(系统)在热解过程的演化规律,采用ReaxFF MD模拟方法,利用GMD-Reax直接模拟了含有98900个原子的柳林烟煤模型的慢速升温过程、恒温裂解过程和二次反应过程;基于所提出的化学反应分层次检索挥发分自由基不同反应的策略,借助VARxMD实现了挥发分自由基不同类别反应数量的计量,着重考察了挥发分自由基被小分子自由基稳定生成焦油和挥发分自由基之间缩聚反应的竞争关系。
本工作的大规模反应分子动力学模拟得到了挥发分自由基参与的被稳定反应和缩聚反应在不同条件下的演化规律,揭示了挥发分自由基的两类反应机制控制着煤热解过程。模拟结果表明煤热解的焦油和焦炭产物收率是这两类反应竞争协调的结果。其中,被稳定反应主要发生在反应前期或较低温阶段,使得煤热解生成大量的焦油产物;挥发分自由基重聚反应发生在高温阶段,使得煤热解结焦剧烈;而在柳林烟煤主要热解的中间温度阶段,被稳定和缩聚化学反应竞争激烈,并会经历从被稳定反应占主导到两类反应竞争激烈,再到缩聚反应占主导三个过程。从被稳定反应占主导到两类化学反应激烈竞争的转折点与焦油产物的收率最高值一致;两类化学反应竞争激烈到缩聚反应占主导的转折点与焦炭产物的起始生成一致。煤热解中挥发分自由基参与的化学反应竞争特性可为工业生产获得高收率焦油,并进一步获得高品质的化学品提供理论指导依据。
参考文献
煤化工在“碳中和”历程中不可或缺
[N].
Coal chemical engineering is indispensable in the process of “carbon neutralization”
[N].
Mild conversion of coal for producing valuable chemicals
[J].
Coal pyrolysis: experiments, kinetic rates and mechanisms
[J].
温度梯度与产物流动对先锋褐煤热解产物分布的影响
[J].
Effects of temperature gradient and product flow on distribution of pyrolysis products of Xianfeng lignite
[J].
煤化学的前沿与挑战: 结构与反应
[J].
Advancement in coal chemistry: structure and reactivity
[J].
Progress in coal pyrolysis
[J].
Behaviors of radical fragments in tar generated from pyrolysis of 4 coals
[J].
Behavior of radicals during solvent extraction of three low rank bituminous coals
[J].
Pyrolysis of coal in TGA: extent of volatile condensation in crucible
[J].
Behaviors of coking and radicals during reaction of volatiles generated from fixed-bed pyrolysis of a lignite and a subbituminous coal
[J].
Focusing on mesoscales: from the energy-minimization multiscale model to mesoscience
[J].
Toward a mesoscale-structure-based kinetic theory for heterogeneous gas-solid flow: particle velocity distribution function
[J].
The utility of coal molecular models
[J].
The molecular representations of coal — a review
[J].
ReaxFF: a reactive force field for hydrocarbons
[J].
The ReaxFF reactive force-field: development, applications and future directions
[J].
Pyrolysis of a large-scale molecular model for Illinois No.6 coal using the ReaxFF reactive force field
[J].
枣泉煤分子模型构建及热解的分子模拟
[J].
Molecular model and pyrolysis simulation of Zaoquan coal
[J].
Early maturation processes in coal(Ⅱ): Reactive dynamics simulations using the ReaxFF reactive force field on Morwell brown coal structures
[J].
Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF
[J].
Effect of calcium on the secondary reactions of tar from Zhundong coal pyrolysis: a molecular dynamics simulation using ReaxFF
[J].
Dynamic migration mechanism of organic oxygen in Fugu coal pyrolysis by large-scale ReaxFF molecular dynamics
[J].
Thermal decomposition process in algaenan of Botryococcus braunii race L(Ⅱ): Molecular dynamics simulations using the ReaxFF reactive force field
[J].
Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field
[J].
Algorithms of GPU-enabled reactive force field (ReaxFF) molecular dynamics
[J].
Dynamic profiles of tar products during Naomaohu coal pyrolysis revealed by large-scale reactive molecular dynamic simulation
[J].
Reaction analysis and visualization of ReaxFF molecular dynamics simulations
[J].
Pyrolysis of Liulin coal simulated by GPU-based ReaxFF MD with cheminformatics analysis
[J].
Pyrolysis simulations of Fugu coal by large-scale ReaxFF molecular dynamics
[J].
Construction of a multicomponent molecular model of Fugu coal for ReaxFF-MD pyrolysis simulation
[J].
Capturing the dynamic profiles of products in Hailaer brown coal pyrolysis with reactive molecular simulations and experiments
[J].
ReaxFF molecular dynamics simulations of thermal reactivity of various fuels in pyrolysis and combustion
[J].
Investigation of model scale effects on coal pyrolysis using ReaxFF MD simulation
[J].
Investigation of overall pyrolysis stages for Liulin bituminous coal by large-scale ReaxFF molecular dynamics
[J].
Initial mechanisms for an overall behavior of lignin pyrolysis through large-scale ReaxFF molecular dynamics simulations
[J].
Initial reactivity of linkages and monomer rings in lignin pyrolysis revealed by ReaxFF molecular dynamics
[J].
Initial reaction mechanisms of cellulose pyrolysis revealed by ReaxFF molecular dynamics
[J].
Application of the ReaxFF reactive force field to reactive dynamics of hydrocarbon chemisorption and decomposition
[J].
ReaxFF MD模拟的物种和化学反应自动分类及可视化
[J].
Automatic classification and visualization of species and reactions obtained from ReaxFF MD simulations
[J].
ReaxFF MD模拟结果分析中化学反应路径网络的发现
[J].
Discovery of chemical reaction networks in analysis of ReaxFF MD simulations
[J].
ReaxFF MD局部区域反应追踪与物理性质可视化分析
[J].
Visualized reaction tracking and physical property analysis for a picked 3D area in a reactive molecular dynamics simulation system
[J].

 京公网安备 11010102001995号
京公网安备 11010102001995号{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}