



引 言
DhaA (EC3.8.1.5)属于烷基卤脱卤酶,能够通过催化碳卤键水解从而有效地降解1-氯丁烷、1,3-二氯丙烷、1,2,3-三氯丙烷以及其他含卤农用杀虫剂[1]。2001年,DhaA被报道用于降解化学毒剂芥子气及其模拟剂,迅速成为了国内外化学毒剂洗消领域的研究热点[2,3,4,5,6]。但DhaA在实际应用中稳定性不高,特别是在尿素、DMSO等变性剂影响下易丧失催化反应能力[7]。随后定向进化、化学修饰、固定化等多种手段均被报道用来提高DhaA的稳定性。Damborsky课题组[8]通过定向进化手段得到了突变体DhaA106,在40%DMSO溶液中的稳定性提高了10倍;Zhao等[9]通过PEG修饰将DhaA在40%DMSO中的催化活性提高了30%。Zheng等[10]通过氨基化介孔泡沫固定化的方式稳定化DhaA,将3 mol/L尿素溶液中放置1 h后的DhaA活性残留率从46.1%提高到71.3%,40%DMSO中放置5 h后的DhaA活性残留率从45.4%提高到85.7%。上述研究虽然能够提高DhaA的稳定性,但对DhaA在尿素和DMSO溶液中的变性过程并未进行探讨,而理解DhaA的变性过程有助于解释其稳定化机理,并为DhaA的进一步稳定化提供指导。前期开展了DhaA在尿素、DMSO溶液中变性过程的荧光光谱研究,发现DhaA在两种变性体系中的变性过程存在明显区别[11]。但由于DhaA变性过程非常迅速,实验技术很难获得DhaA的结构变化信息,无法从原子和分子角度解释其变性过程的差异。
分子动力学(MD)模拟能够利用计算机构建的分子模型模拟蛋白的微观结构和动态行为,目前已经广泛用于蛋白质折叠和去折叠过程研究,MD模拟具有更高维度的动力学和更高分辨率的结构分析能力,逐渐成为了微观层面研究蛋白变性机理的重要工具[12,13,14,15,16,17,18]。Roccatano等[19]在纯水和DMSO水溶液对细胞色素P450 BM-3进行了MD模拟,发现14% DMSO溶液中BM-3的底物进出口通道出现较大变化,DMSO分子出现在底物结合位点附近,导致BM-3变性。Li等[20]采用MD模拟了谷胱甘肽S转移酶在不同温度尿素溶液中的变性过程。Khan等[21]采用实验方法和MD模拟相结合的方式研究了细胞色素c在尿素和盐酸胍中的变性过程,发现在盐酸胍诱导下细胞色素c的变性过程中存在折叠态、去折叠态以外的中间态。沈洪辰等[22]通过MD模拟了蛋白p35突变体p35C的构象变化,对突变后结构稳定性增强的机理进行了分析。卢滇楠等[23,24]和潘晓莉等[25]同样采用MD模拟,分别研究了表面活性剂、二硫键以及离子液体对蛋白质稳定性的影响。
本文以DhaA为研究对象,采用全原子MD模拟方法,考察了DhaA在尿素和DMSO溶液中的变性情况,通过分析DhaA的结构变化和其与溶剂分子的相互作用,研究DhaA在两种体系中的变性过程。
1 模型和方法
1.1 模拟模型
图1
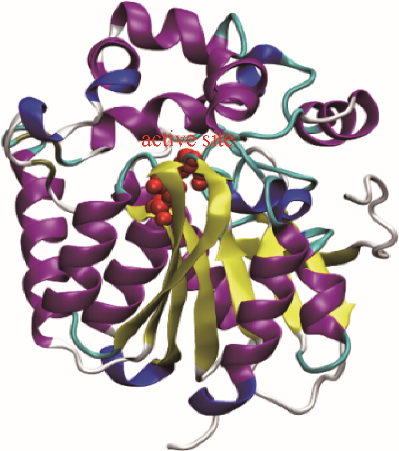
图1
DhaA的三维晶体结构(红色珠子代表活性位点)
Fig.1
Native three dimensional structure of DhaA(red beads represent catalytic sites)
1.2 模拟方法
模拟盒子的尺寸为8 nm×8 nm×8 nm,水分子模型采用TIP3P模型。DhaA的所有酸性残基(包括谷氨酸和天冬氨酸)和C-端残基均进行去质子化处理,而所有碱性残基(包括赖氨酸和精氨酸)以及N-端残基均进行质子化处理。DhaA带有-17单位净电荷。为了保持体系呈电中性,体系中加入相应的反号离子(Na+),在x,y,z三个方向施加周期性边界条件。在MD模拟之前,采用最陡下降法对体系进行能量最小化处理,以消除体系中不合理的原子空间位置重叠或者不适当的几何结构。之后在NVT条件下模拟运行100 ps进行温度预平衡,再在NPT条件下模拟100 ps进行压力预平衡,模拟步长为2 fs。温度控制采用Nose-Hoover恒温器,时间常数为0.5 ps。压力控制采用Parrinello-Rahman恒压器,时间常数为2 ps。体系的模拟温度为298 K,压力为1×105 Pa。体系中所有包含氢原子的化学键都用LINCS算法限制,非键相互作用截断半径为1.0 nm,静电作用的计算使用PME方法,范德华作用的计算采用switch方法,在0.9~1.0 nm之间势能平滑地过渡到0。最后所有的体系都在NPT系综中进行MD模拟,所有体系的模拟时间均为1000 ns。模拟体系为5.6 mol/L 尿素和DMSO溶液(加入1500个尿素或DMSO分子),纯水溶液作为对照。所有MD模拟计算均在华南理工大学高性能网格计算中心SCUTGrid服务器 (含20 个节点的8 核Intel Xeon E-2670 CPU和64G 内存)上完成。
2 实验结果与讨论
2.1 DhaA结构分析
DhaA主链结构的均方根偏差(root-mean-square deviation,RMSD)表示了DhaA变性过程中结构变化的最小均方根偏差,能够定量地表征DhaA结构变化大小。图2为DhaA在水溶液、5.6 mol/L尿素及DMSO三种体系中蛋白骨架的RMSD值。可以看到,由于DhaA的结构稳定性不高,在纯水模拟体系中存在一定程度的结构变化,RMSD平均值为(0.17±0.03) nm。DhaA在5.6 mol/L 尿素体系模拟过程中,RMSD值在431 ns左右出现较大波动,峰值达到0.5 nm,而RMSD的平均值为(0.21±0.03) nm。DMSO同样会造成DhaA的结构发生波动,在5.6 mol/L DMSO溶液中DhaA的RMSD值在887 ns达到0.4 nm左右,RMSD平均值为(0.20±0.04) nm。比较三种体系中DhaA的RMSD值可以看到,DhaA在尿素和DMSO中的结构变化程度高于纯水溶液中的,相对变化提高17.6%~23.5%。说明尿素和DMSO两种有机小分子均会诱导DhaA发生一定程度的结构变化。
图2
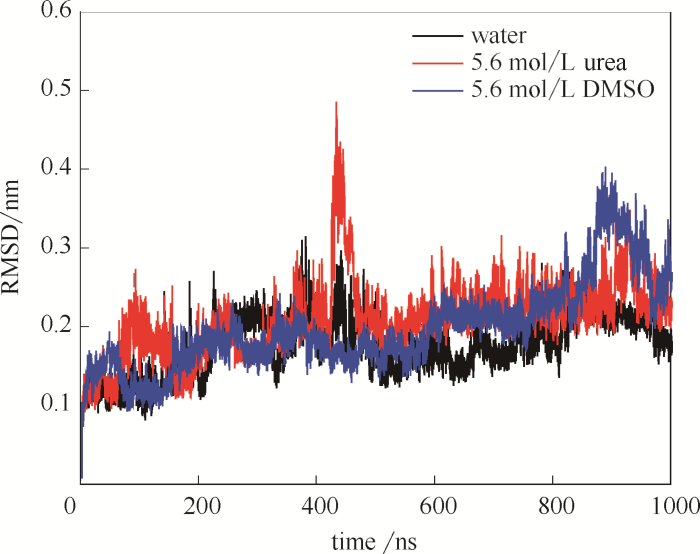
图2
DhaA在不同模拟体系中的RMSD随时间变化
Fig.2
RMSD evolutions of DhaA as a function of time in different simulation systems
图3(a)~(c)显示的是不同模拟体系中,DhaA二级结构之间发生相互转换的动态过程(DSSP)。在整个MD模拟过程中,DhaA的主要二级结构(α-螺旋和β-折叠)几乎没有发生变化,说明DhaA在三种体系在1000 ns模拟过程中二级结构整体保持稳定。
图3
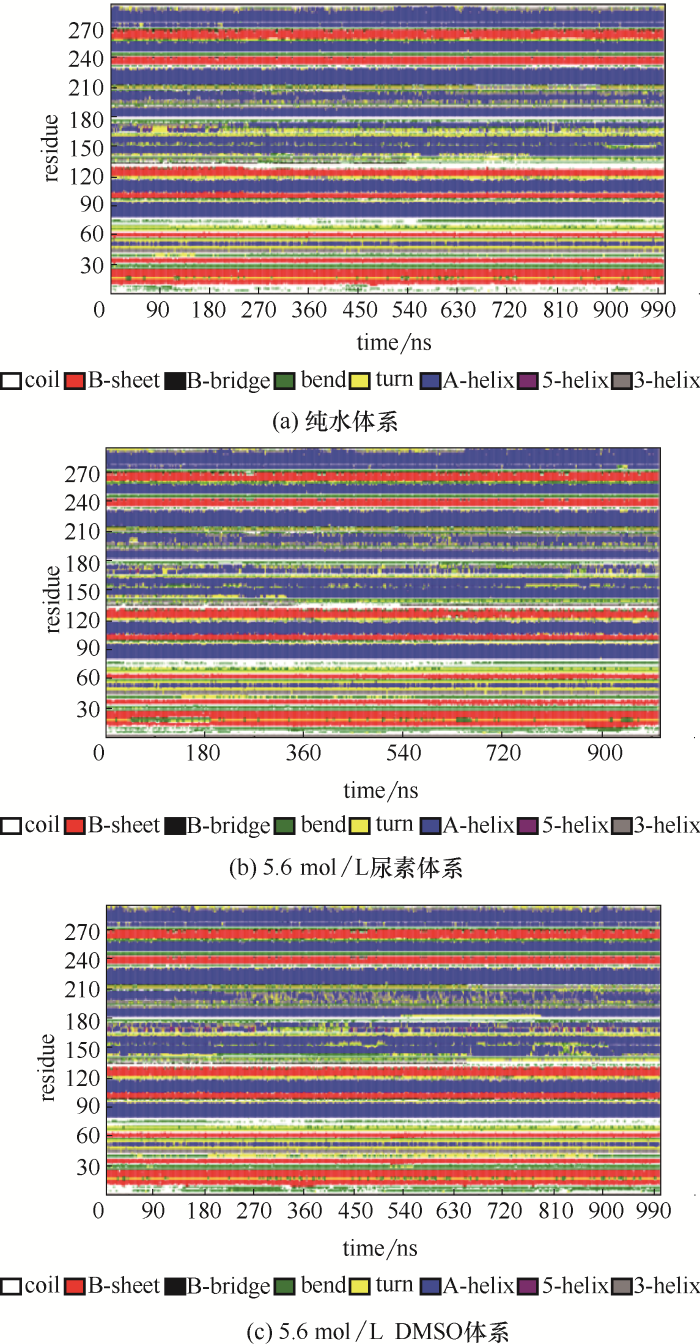
图3
DhaA在不同模拟体系中的二级结构变化
Fig.3
Secondary structure evolutions of DhaA as a function of time in different simulation systems
图4
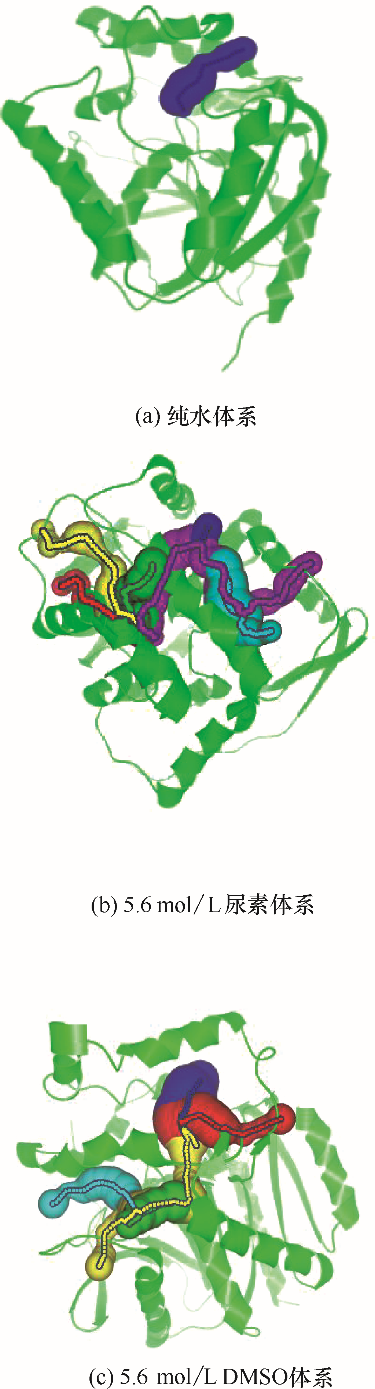
图4
DhaA在不同模拟体系中的通道示意图
Fig.4
Snapshots of tunnel of DhaA in different simulation systems
表1 DhaA在不同变性条件下的主通道结构参数
Table 1
| DhaA模拟体系 | 通道长度/Å | 通道曲率 | 瓶颈尺寸/Å | 通过成本 |
|---|---|---|---|---|
| 纯水 | 12.55 | 1.25 | 1.16 | 0.45 |
| 尿素 | 22.48 | 1.45 | 1.10 | 0.54 |
| DMSO | 10.27 | 1.27 | 1.26 | 0.19 |
从图4(a)~(c)可以看到,DhaA在纯水体系中仅存在1条底物进出口通道,当模拟体系中加入尿素或DMSO分子后,底物进出口通道发生明显变化。在尿素体系中,DhaA的底物进出口通道增加到6条,而在DMSO体系中,底物进出口通道也增加为5条。与此同时,底物进出主通道的结构参数也发生了较大改变。在纯水体系中,DhaA的主通道长度12.55 Å(1 Å=0.1 nm,后同),曲率1.25,通道的瓶颈尺寸为1.16 Å,此时底物在该通道中的通过成本为0.45。当体系中加入尿素分子后,由于尿素分子与DhaA的相互作用,导致DhaA的结构发生变化,主通道长度从12.55 Å增加到22.48 Å,通道曲率也从1.25增加到1.45,瓶颈尺寸从1.16 Å减小到1.10 Å,底物在主通道中的运动受限,通过成本增加为0.54。在DMSO体系中,由于DMSO的作用影响,主通道长度缩短(10.27 Å)、瓶颈尺寸增大(1.26 Å),通道曲率基本得到保持。上述结构变化有利于催化过程中的底物传输,造成底物通过成本降低为0.19。因此可以推测,与尿素分子结合后,DhaA通道结构变得更加紧密,底物通过难度增加;而与DMSO分子结合后,DhaA通道结构变得松散,底物很容易进出,这是尿素、DMSO两种分子与DhaA作用方式不同造成的。
2.2 有机小分子与DhaA作用分析
通过分析DhaA在不同体系中氢键(设定给体和受体之间的距离截断值为3.5 Å,给体-供体-氢原子之间的角度截断值为30°)数目的变化,可以比较尿素和DMSO分子对DhaA变性过程的影响。如图5所示,在纯水溶液中,DhaA蛋白内存在200个左右氢键,蛋白与水分子的氢键数量在600个左右。当体系中存在尿素分子时,DhaA与水分子的氢键减少到500个左右,缺少的氢键由DhaA与尿素分子形成的氢键进行了补充,体系氢键总数增加。说明尿素分子形成氢键的能力高于水分子,尿素分子能够取代DhaA周围的水分子,通过氢键与DhaA结合。
图5
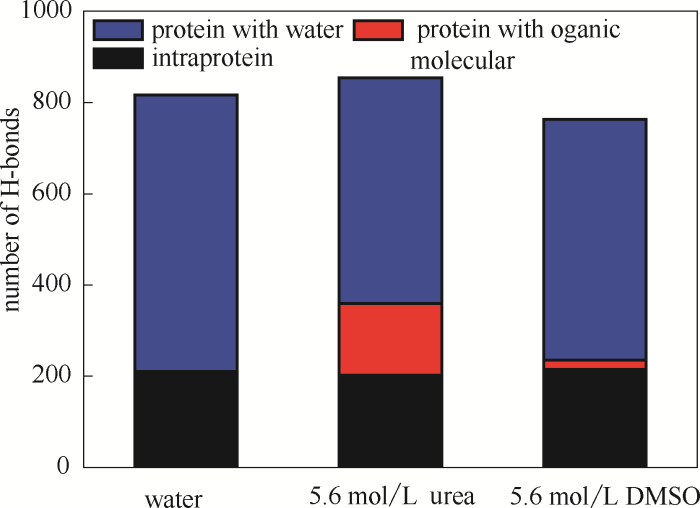
图5
DhaA在不同模拟体系中的氢键数(蓝色表示DhaA与水分子氢键,红色表示DhaA与有机分子氢键,黑色表示DhaA分子内氢键)
Fig.5
Numbers of H-bonds in DhaA in simulation systems
(blue bar represents H-bond between DhaA and water molecules, red bar represents H-bond between DhaA and organic molecules, black bar represents intramolecular H-bond of DhaA molecules)
在DMSO体系中,DhaA分子内氢键与纯水溶液中一致,DhaA与水分子形成的氢键数目仍然减少至500个左右,但DhaA与DMSO分子仅仅形成了20个氢键,体系氢键总数减少。说明DMSO分子与DhaA形成氢键的能力不强,氢键总数的减少和DMSO体系中DhaA的构象变化有关。
DhaA的催化活性中心位于疏水空腔内,在DhaA变性过程中,有机小分子可以进入疏水空腔与活性位点结合,从而导致DhaA活性衰减和构象变化。不同体系中,距离催化活性位点3.5 Å范围内的溶剂分子分布如图6所示。
图6
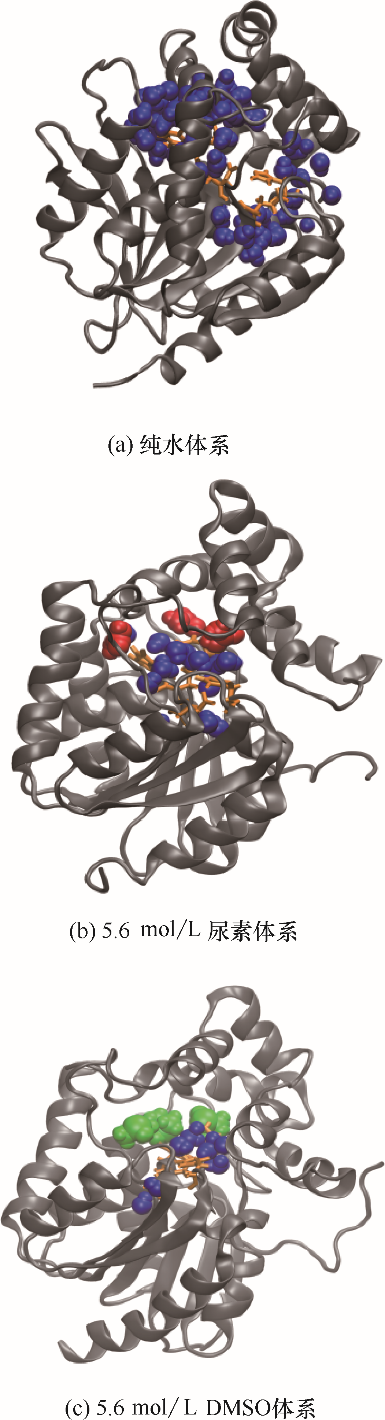
图6
DhaA在不同模拟体系中的催化位点3.5 Å内溶剂分子分布(蓝色珠子代表水分子,红色珠子代表尿素分子,绿色珠子代表DMSO分子)
Fig.6
Snapshots of solution molecules distribution within 3.5 Å around catalytic sites in DhaA in different simulation systems
(blue beads represent water molecules, red beads represent urea molecules, green beads represent DMSO molecules)
图7
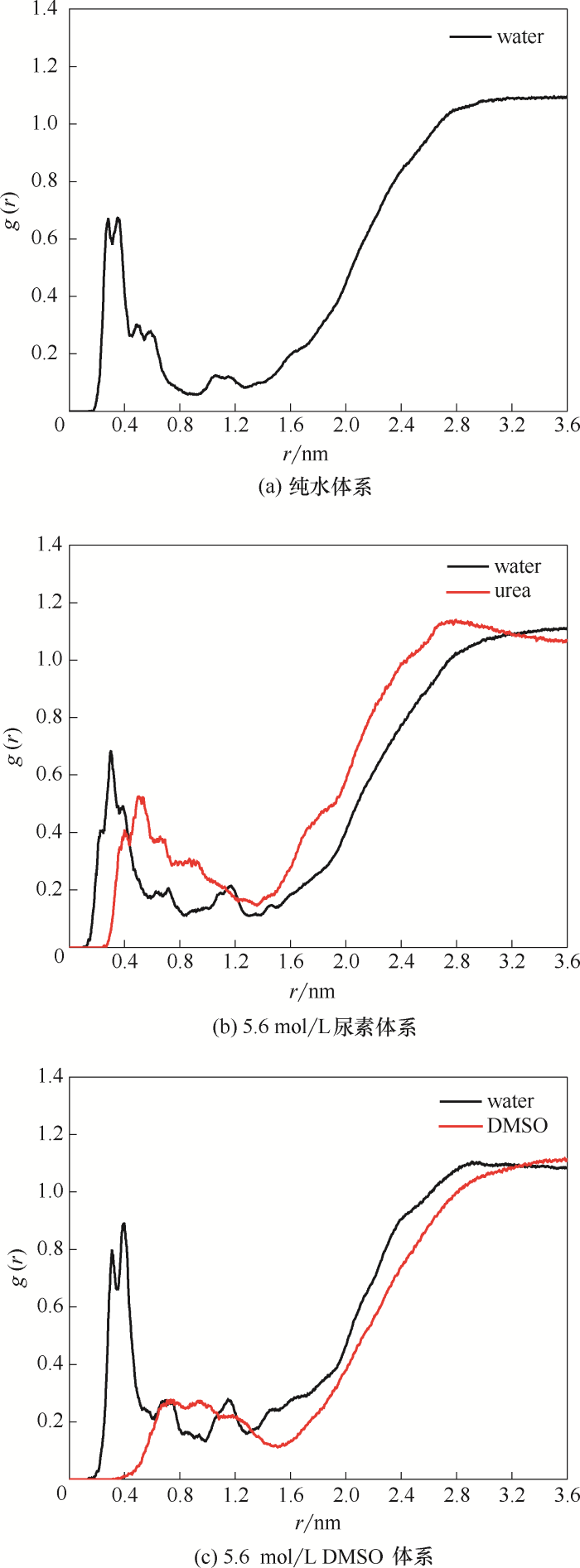
图7
DhaA催化位点附近溶剂分子的径向分布函数
Fig.7
Radial distribution functions (RDFs) of solvent molecules around catalytic sites
DhaA催化位点附近溶剂分子的分布主要集中在0~1.2 nm和2~3.5 nm两个区域,其中0~1.2 nm区域的分布比较典型。在纯水体系中[图7(a)],0.4 nm处存在一个很突出的尖峰,此处水分子通过氢键作用与催化位点结合,同样在0.6 nm处也存在一个小峰。图7(b)描述了5.6 mol/L尿素体系中尿素分子和水分子在DhaA催化位点附近的分布。可以看到,体系中尿素分子跟水分子的分布情况相似,说明溶液中尿素分子能被水分子很好溶解。虽然0.4 nm处的水分子分布与纯水体系中相差不大,但开始出现部分尿素分子,而0.6 nm处尿素分子的分布函数高于水分子,说明尿素分子进入DhaA催化位点附近,并且开始取代附近的水分子。在图7(c)所示的5.6 mol/L DMSO体系中,0.4 nm处水分子分布得到增强,说明DMSO加入后无法通过氢键作用结合催化位点,反而增强了水分子与催化位点之间的氢键作用。但在催化位点附近0.6 nm处以及水分子分布较少0.8 nm处,DMSO分布逐渐增多,此处分布有DhaA的疏水氨基酸,DMSO能够通过范德华作用稳定存在。
尿素和DMSO分子与催化位点的相互作用,同样可以通过研究三种模拟体系中DhaA催化位点的结构变化来进行验证。对比DhaA在纯水和5.6 mol/L尿素体系中的局部结构[图8(a)],可以发现,由于尿素分子与各催化残基之间存在氢键作用,导致催化位点Asn41、Asp106、Trp107、Glu130和His272均发生了明显的位置变化。而在5.6 mol/L DMSO体系中,DMSO分子只能与非极性的Trp107发生范德华作用,造成Asp106-Trp107结构发生位移,其他催化位点结构没有明显变化。
图8
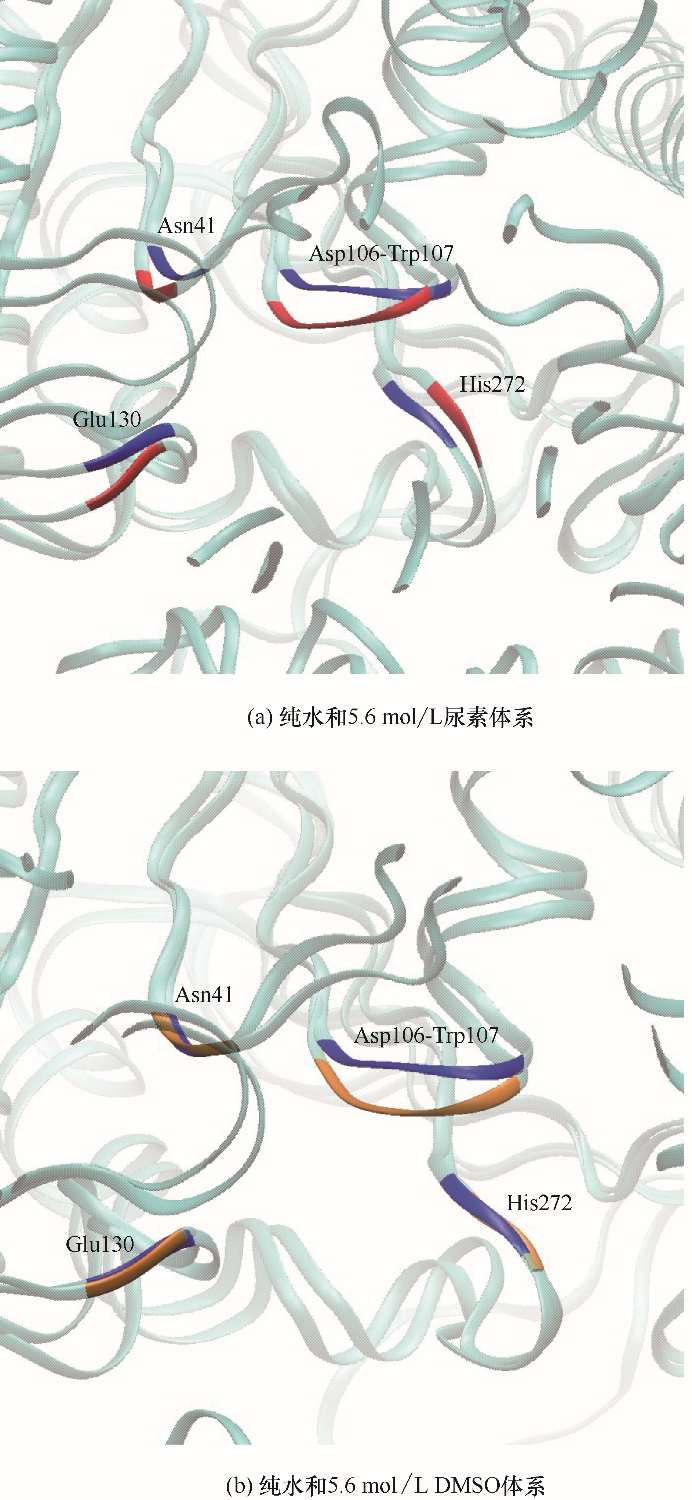
图8
DhaA在模拟体系中催化位点的结构变化(蓝色为纯水体系中的DhaA,红色为尿素体系中的DhaA,橙色为DMSO体系中的DhaA)
Fig.8
Structural change of catalytic sites of DhaA in different simulation systems(blue part represents DhaA in water, red part represents DhaA in urea solution, orange part represents DhaA in DMSO solution)
2.3 DhaA变性过程分析
通过前面分析可知,在DhaA变性过程中,尿素分子能够通过氢键作用取代DhaA外层的水分子,从而在DhaA分子周围富集,并且能够进入DhaA催化位点附近,与催化位点形成氢键。大量尿素分子与DhaA的相互作用造成DhaA底物进出口通道的通道长度增加、瓶颈尺寸减小,导致DhaA变性。DMSO诱导DhaA变性过程中,DMSO分子无法取代水分子与DhaA形成氢键,但部分DMSO分子能够通过范德华作用进入DhaA疏水空腔,造成其疏水结构暴露,引起DhaA底物进出口通道长度缩短、瓶颈尺寸增加,诱导DhaA发生变性。
图9
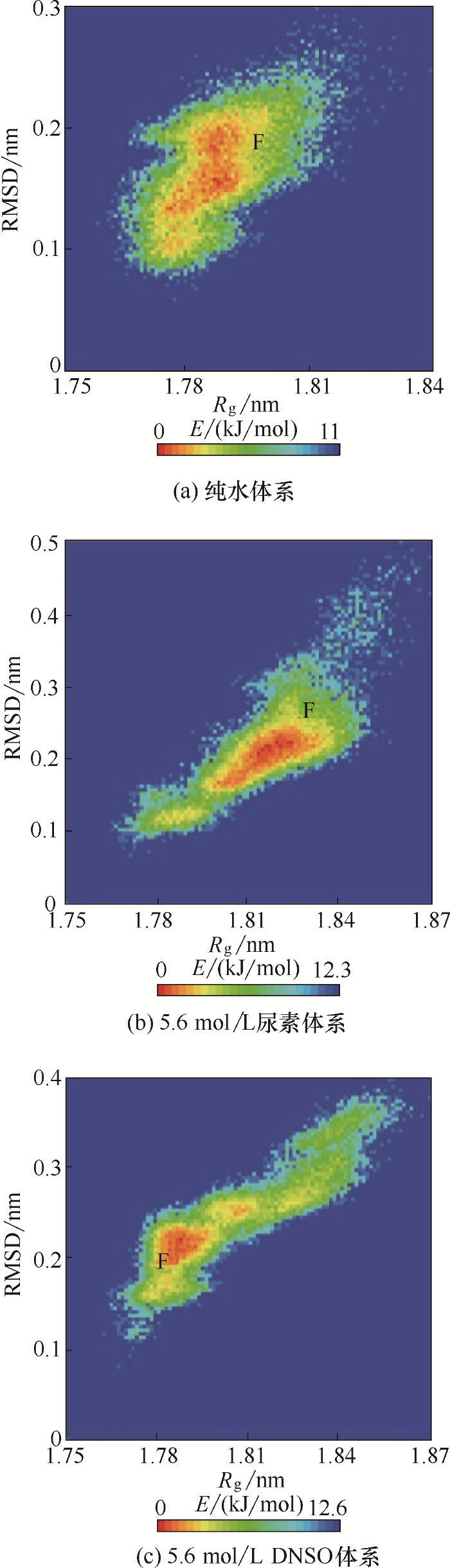
图9
DhaA在不同模拟体系中的自由能形貌图(F表示折叠态)
Fig.9
Gibbs free energies landscapes of DhaA in different simulation systems (F indicated folding state)
3 结 论
本文通过MD模拟研究了DhaA在尿素和DMSO两种溶液体系中的变性过程。模拟结果显示,虽然DhaA在三种体系中RMSD和DSSP变化不大,但底物进出口通道结构变化明显。在5.6 mol/L尿素体系中,尿素分子能够取代DhaA周围水分子,造成DhaA主通道长度增加、通道曲率增大、瓶颈尺寸减小,底物进出难度增加;而在5.6 mol/L DMSO体系中,DMSO分子进入疏水空腔,诱导DhaA发生构象变化,使得通道长度缩短、瓶颈尺寸增大,底物更容易进出。与此同时,溶剂分子的径向分布函数证实,尿素分子能够进入DhaA催化位点附近,并通过氢键作用与位点结合,而DMSO仅能通过范德华作用稳定存在于DhaA催化位点附近。自由能形貌图表明,由于DMSO造成DhaA构象变化,导致其构象分布更广,并且存在其他稳定构象。DMSO体系中其他稳定构象的验证和鉴定,需要采用分子动力学模拟结合实验进一步研究。掌握DhaA的上述变性过程有助于深入研究各种稳定化手段的作用机理,为DhaA稳定性的进一步提高提供理论指导。
参考文献
Haloalkane dehalogenases: biotechnological applications
[J].
Properties and biotechnological applications of natural and engineered haloalkane dehalogenases
[J].
Enzymatic degradation of HD
[R].
Enzyme-based test strips for visual or photographic detection and quantitation of gaseous sulfur mustard
[J].
烷基卤去卤化酶对芥子气的催化水解
[J].
Catalytic hydrolysis of sulfur mustard by haloalkane dehalogenases
[J].
多点突变提高DhaA对芥子气的活性和热稳定性
[J].
Improvement in the thermostability and activity of DhaA against sulfur mustard by multipoint mutagenesis
[J].
Organic co-solvents affect activity, stability and enantioselectivity of haloalkane dehalogenases
[J].
Balancing the stability-activity trade-off by fine-tuning dehalogenase access tunnels
[J].
PEGylation with the thiosuccinimido butylamine linker significantly increases the stability of haloalkane dehalogenase DhaA
[J].
Mesoporous support designed for DhaA adsorption with improved stability
[J].
荧光光谱法研究氨基改性介孔泡沫对DhaA的稳定化机理
[J].
Stabilization mechanism of amino-mesocellular foam to DhaA by fluorescence spectroscopic method
[J].
Molecular dynamics simulations of the protein unfolding/folding reaction
[J].
Simplicity within the complexity: bilateral impact of DMSO on the functional and unfolding patterns of α-chymotrypsin
[J].
Protein folding: molecular dynamics simulations and in vitro studies for probing mechanism of urea- and guanidinium chloride induced unfolding of horse cytochrome-c
[J].
Characterization of mechanical unfolding intermediates of membrane proteins by coarse grained molecular dynamics simulation
[J].
Cosolvent effects on protein stability
[J].
β发卡多肽Trpzip4折叠的副本交换分子动力学模拟
[J].
Replica exchange molecular dynamics simulations on the folding of Trpzip4 β-hairpin
[J].
分子动力学模拟技术在生物分子研究中的进展
[J].
Recent developments in using molecular dynamics simulation techniques to study biomolecules
[J].
Structural and dynamic properties of cytochrome P450 BM-3 in pure water and in a dimethylsulfoxide/water mixture
[J].
Thermal and urea induced unfolding processes of glutathione S-transferase by molecular dynamics simulation
[J].
Structural basis of urea-induced unfolding: unraveling the folding pathway of hemochromatosis factor E
[J].
Y220C突变体影响p53C蛋白质构象转换的分子动力学模拟
[J].
Effect of Y220C mutant on the conformational transition of p53C probed by molecular dynamics simulation
[J].
蛋白质-表面活性剂组装结构的分子模拟
[J].
Molecular simulation of protein-surfactant assembly in aqueous solution
[J].
分子动力学模拟二硫键对胰岛素构象稳定性的影响
[J].
Molecular dynamics simulation of impact of disulfide bridge on conformational stability of insulin
[J].
胰岛素活性结构在水合离子液体中的稳定性
[J].
Bioactive structural stability of insulin in hydrated ionic liquids
[J].
GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation
[J].
脱卤酶对芥子气的催化活性和稳定性研究
[D].
Study on the activity and stability of dehalogenase against sulfur mustard
[D].
CAVER Analyst 2.0: analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories
[J].
Elucidation of stable intermediates in urea induced unfolding pathway of human carbonic anhydrase Ⅸ
[J].

 京公网安备 11010102001995号
京公网安备 11010102001995号{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}