CIESC Journal ›› 2019, Vol. 70 ›› Issue (7): 2540-2547.DOI: 10.11949/0438-1157.20190065
• Catalysis, kinetics and reactors • Previous Articles Next Articles
Nan SONG( ),Minjian PAN,Bingxu CHEN,Gang QIAN,Xuezhi DUAN(),Xinggui ZHOU
),Minjian PAN,Bingxu CHEN,Gang QIAN,Xuezhi DUAN(),Xinggui ZHOU
Received:2019-01-21
Revised:2019-04-04
Online:2019-07-05
Published:2019-07-05
Contact:
Xuezhi DUAN
宋楠(),潘敏建,陈炳旭,钱刚,段学志(),周兴贵
通讯作者:
段学志
作者简介:宋楠(1983—),女,博士研究生,讲师,<email>cuiky@ecust.edu.cn</email>
基金资助:CLC Number:
Nan SONG, Minjian PAN, Bingxu CHEN, Gang QIAN, Xuezhi DUAN, Xinggui ZHOU. CH4 formation and C—C coupling mechanism on (011) surface of η-Fe2C Fischer-Tropsch catalyst[J]. CIESC Journal, 2019, 70(7): 2540-2547.
宋楠, 潘敏建, 陈炳旭, 钱刚, 段学志, 周兴贵. 费托催化剂η-Fe2C (011)上CH4形成及C-C耦合机理研究[J]. 化工学报, 2019, 70(7): 2540-2547.
Add to citation manager EndNote|Ris|BibTeX

Fig.1 Top and side views of favorable adsorption configurations of H (a) and CHx (b) on η-Fe2C(011) surface and corresponding top and side views of TSs of elementary steps involved in methanation (c)(blue: Fe atoms; grey: C atoms of iron carbide; green: C atoms involved in reactions; white: H atoms; yellow: H atoms involved in reactions)
| Site | rFe-H / nm | rC-H / nm | Eads /eV |
|---|---|---|---|
| 2F | 0.172,0.170 | -2.22 | |
| 3F | 0.170,0.178,0.184 | -2.27 | |
| 4F | 0.112 | -2.03 |
Table 1 Key energetics and structural parameters of identified H adsorption on perfect η-Fe2C(011) surface
| Site | rFe-H / nm | rC-H / nm | Eads /eV |
|---|---|---|---|
| 2F | 0.172,0.170 | -2.22 | |
| 3F | 0.170,0.178,0.184 | -2.27 | |
| 4F | 0.112 | -2.03 |
| Reaction | dC—H /nm | Ea /eV | ΔrE /eV |
|---|---|---|---|
| C+ H | 0.142 | 0.49 (0.44) | 0.15 (0.19) |
| CH + H | 0.147 | 0.93 (0.83) | 0.44 (0.50) |
| CH2 + H | 0.160 | 0.27 (0.25) | -0.41 (-0.31) |
| CH3+ H | 0.157 | 0.56 (0.56) | -0.18 (0.08) |
Table 2 C—H distances (dC—H) at TSs, reaction barriers (Ea) and reaction energies (ΔrE) involved in CH4 formation on η-Fe2C(011) surface (values including ZPE in parentheses)
| Reaction | dC—H /nm | Ea /eV | ΔrE /eV |
|---|---|---|---|
| C+ H | 0.142 | 0.49 (0.44) | 0.15 (0.19) |
| CH + H | 0.147 | 0.93 (0.83) | 0.44 (0.50) |
| CH2 + H | 0.160 | 0.27 (0.25) | -0.41 (-0.31) |
| CH3+ H | 0.157 | 0.56 (0.56) | -0.18 (0.08) |
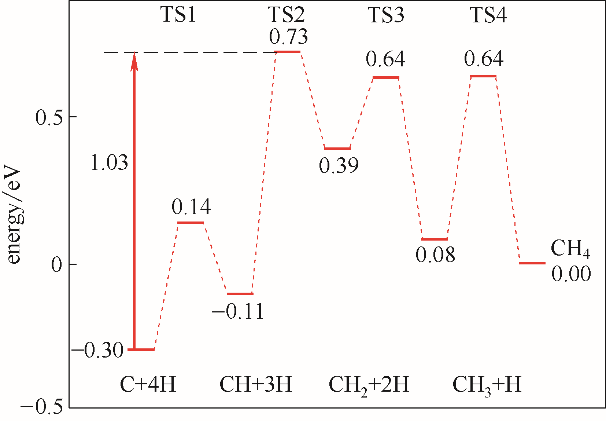
Fig.2 Energy profiles of CH4 formation on η-Fe2C(011) surface (corresponding effective barriers were also presented)
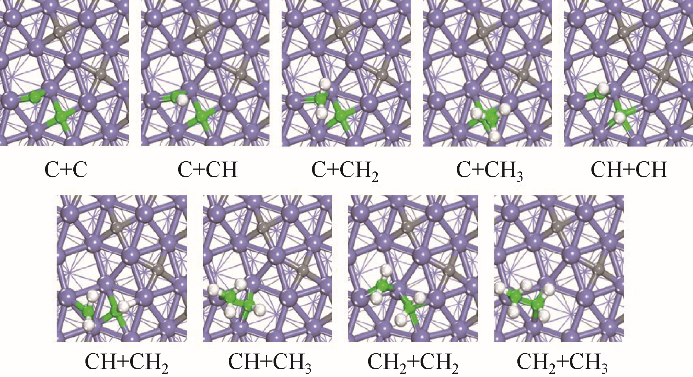
Fig.3 Structures of TSs of C—C coupling reactions
| Reaction | dC-C/nm | Ea/eV | ΔrE/eV | |
|---|---|---|---|---|
| C+C | 0.222 | 3.00 (2.98) | 1.15 (1.15) | 3.00 (2.98) |
| C+CH | 0.224 | 1.70 (1.74) | 0.56 (0.60) | 1.85 (1.93) |
| C+CH2 | 0.201 | 1.17 (1.18) | 0.33 (0.38) | 1.76 (1.87) |
| C+CH3 | 0.197 | 1.14 (1.14) | 0.54 (0.58) | 1.32 (1.52) |
| CH+CH | 0.192 | 1.89 (1.90) | 0.62 (0.68) | 2.18 (2.29) |
| CH+CH2 | 0.206 | 1.22 (1.19) | 0.54 (0.57) | 1.95 (2.08) |
| CH+CH3 | 0.203 | 1.60 (1.63) | 0.80 (0.84) | 1.92 (2.21) |
| CH2+CH2 | 0.215 | 0.96 (0.98) | -0.02 (0.09) | 2.13(2.36) |
| CH2+CH3 | 0.203 | 1.19 (1.27) | 0.42 (0.57) | 1.95 (2.34) |
Table 3 C-C distances (dC-C) at TSs and reaction barriers (Ea), reaction energies (ΔrE) and effective activation energies (Eeff,CHi-CHj) of C1+C1 coupling reactions on η-Fe2C(011) surface (values including ZPE in parentheses)
| Reaction | dC-C/nm | Ea/eV | ΔrE/eV | |
|---|---|---|---|---|
| C+C | 0.222 | 3.00 (2.98) | 1.15 (1.15) | 3.00 (2.98) |
| C+CH | 0.224 | 1.70 (1.74) | 0.56 (0.60) | 1.85 (1.93) |
| C+CH2 | 0.201 | 1.17 (1.18) | 0.33 (0.38) | 1.76 (1.87) |
| C+CH3 | 0.197 | 1.14 (1.14) | 0.54 (0.58) | 1.32 (1.52) |
| CH+CH | 0.192 | 1.89 (1.90) | 0.62 (0.68) | 2.18 (2.29) |
| CH+CH2 | 0.206 | 1.22 (1.19) | 0.54 (0.57) | 1.95 (2.08) |
| CH+CH3 | 0.203 | 1.60 (1.63) | 0.80 (0.84) | 1.92 (2.21) |
| CH2+CH2 | 0.215 | 0.96 (0.98) | -0.02 (0.09) | 2.13(2.36) |
| CH2+CH3 | 0.203 | 1.19 (1.27) | 0.42 (0.57) | 1.95 (2.34) |
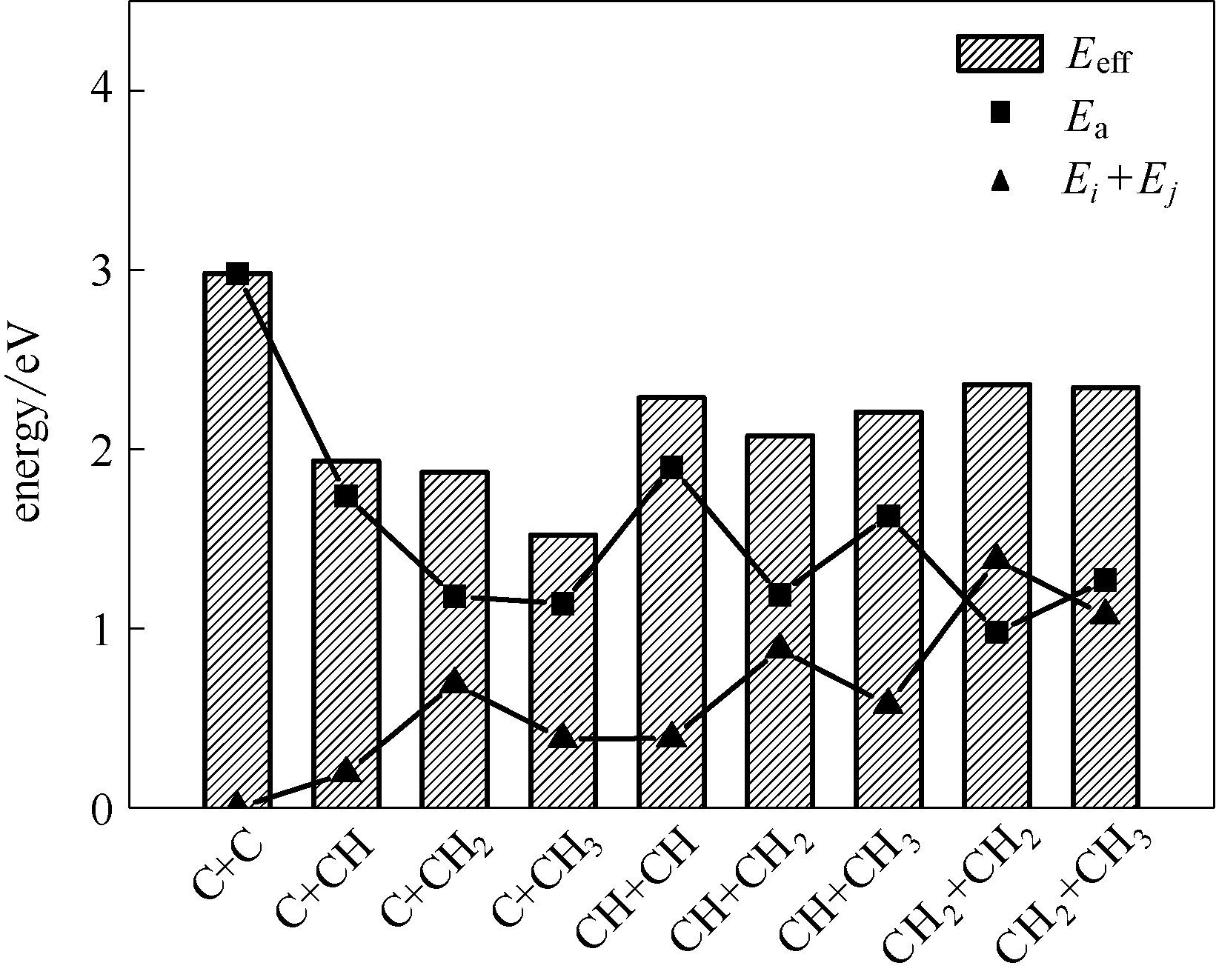
Fig.4 Barriers, relative energy of reactants and effective barriers of CHi+CHj coupling on η-Fe2C(011) surface
| Surface | The most possible C—C coupling route | Ea/eV | Ref. | |
|---|---|---|---|---|
Fe | (210) | C+CH3 | 1.10 | [38] |
(100) | C+CH2 C+CH3 | 0.73 | [31] [31] | |
| 0.49 | ||||
χ-Fe5C2 | (100) | C+CH3 | 1.02 | [26] |
| (001) | C+CO | 0.66 | [39] | |
(510) | C+CH CH+CH | 1.09 | [16] [16] | |
| 0.96 | ||||
θ-Fe3C | (031) | CH+CH CH2+CH2 | 0.76 | [17] [17] |
| 0.50 | ||||
η-Fe2C | (011) | C+CH3 CH2+CH2 | 1.14 | this work this work |
| 0.98 | ||||
Table 4 Most possible routes and their activation barriers of C1+C1 coupling on different iron carbide surfaces
| Surface | The most possible C—C coupling route | Ea/eV | Ref. | |
|---|---|---|---|---|
Fe | (210) | C+CH3 | 1.10 | [38] |
(100) | C+CH2 C+CH3 | 0.73 | [31] [31] | |
| 0.49 | ||||
χ-Fe5C2 | (100) | C+CH3 | 1.02 | [26] |
| (001) | C+CO | 0.66 | [39] | |
(510) | C+CH CH+CH | 1.09 | [16] [16] | |
| 0.96 | ||||
θ-Fe3C | (031) | CH+CH CH2+CH2 | 0.76 | [17] [17] |
| 0.50 | ||||
η-Fe2C | (011) | C+CH3 CH2+CH2 | 1.14 | this work this work |
| 0.98 | ||||
| Surface | ΔEeff/eV | Site | Ref. | ||
|---|---|---|---|---|---|
| Fe(210) | 2.13 | 2.19 | -0.06 | step | [28] |
| Fe(100) | 2.13 | 1.92 | 0.21 | step | [24-45] |
| χ-Fe5C2(100) | 1.89 | 1.94 | -0.05 | step | [26] |
| χ-Fe5C2(510) | 2.39 | 1.66 | 0.73 | terrace | [16] |
| θ-Fe3C(031) | 2.29 | 0.99 | 1.30 | terrace | [17] |
| η-Fe2C(011) | 1.03 | 1.52 | -0.49 | step | this work |
Table 5 Effective barriers of CH4 formation and C1+C1 coupling and their barrier differences on different iron carbide surfaces
| Surface | ΔEeff/eV | Site | Ref. | ||
|---|---|---|---|---|---|
| Fe(210) | 2.13 | 2.19 | -0.06 | step | [28] |
| Fe(100) | 2.13 | 1.92 | 0.21 | step | [24-45] |
| χ-Fe5C2(100) | 1.89 | 1.94 | -0.05 | step | [26] |
| χ-Fe5C2(510) | 2.39 | 1.66 | 0.73 | terrace | [16] |
| θ-Fe3C(031) | 2.29 | 0.99 | 1.30 | terrace | [17] |
| η-Fe2C(011) | 1.03 | 1.52 | -0.49 | step | this work |
| 1 | ZhangQ, KangJ, WangY. Development of novel catalysts for Fischer-Tropsch synthesis: tuning the product selectivity[J]. ChemCatChem, 2010, 2(9): 1030-1058. |
| 2 | 王野, 成康, 张庆红. 一氧化碳加氢制碳氢化合物反应选择性的调控[J]. 中国科学: 化学, 2012, 42(4): 263-375. |
| WangY, ChengK, ZhangQ H. Selectivity tuning for the hydrogenation of carbon monoxide into hydrocarbons[J]. Scientia Sinica Chimica, 2012, 42(4): 263-375. | |
| 3 | JagerB, EspinozaR. Advances in low temperature Fischer-Tropsch synthesis[J]. Catalysis Today, 1995, 23: 17-28. |
| 4 | XuJ, YangY, LiY W. Fischer-Tropsch synthesis process development: steps from fundamentals to industrial practices[J]. Current Opinion in Chemical Engineering, 2013, 2: 354-362. |
| 5 | JahangiriH, BennettJ, MahjoubiP, et al. A review of advanced catalyst development for Fischer-Tropsch synthesis of hydrocarbons from biomass derived syngas[J]. Catalysis Science & Technology, 2014, 4(8): 2210-2229. |
| 6 | SchulzH. Short history and present trends of Fischer-Tropsch synthesis[J]. Applied Catalysis A: General, 1999, 186: 3-12. |
| 7 | 温晓东, 杨勇, 李永旺, 等. 费托合成铁基催化剂的设计基础: 从理论走向实践[J]. 中国科学: 化学, 2017, (11): 72-85. |
| WenX D, YangY, LiY W, et al. The design principle of iron-based catalysts for Fischer-Tropsch synthesis: from theory to practice[J]. Scientia Sinica Chimica, 2017, (11): 72-85. | |
| 8 | de SmitE, CinquiniF, WeckhuysenB M, et al. Stability and reactivity of ϵ-χ-θ iron carbide catalyst phases in Fischer-Tropsch synthesis: controlling μC[J]. Journal of the American Chemical Society, 2010, 132: 14928-14941. |
| 9 | XuK, SunB, QiaoM H, et al. ε-Iron carbide as a low-temperature Fischer-Tropsch synthesis catalyst[J]. Nature Communication, 2014, 5: 5783-5790. |
| 10 | LiuX W, ZhaoS, MengY. Mössbauer spectroscopy of iron carbides: from prediction to experimental confirmation[J]. Scientific Reports, 2016, 6: 26184. |
| 11 | JoséG R C, MaartenK S , Marie-FrancoiseR. First principle study on the adsorption of hdrocarbon chains involved in Fischer-Tropsch synthesis over iron carbides[J]. The Journal of Physical Chemistry C, 2017, 121: 25052-25063. |
| 12 | Le CaerG, DuboisJ M, BussiereP, et al. Characterization by Mossbauer spectroscopy of iron carbides formed by Fischer-Tropsch synthesis[J]. The Journal of Physical Chemistry, 1982, 86(24): 4799-4808. |
| 13 | FangC M, SluiterM H F, van HuisM A, et al. Origin of predominance of cementite among iron carbides in steel at elevated temperature[J]. Physical Review Letters, 2010, 105(5): 055503. |
| 14 | BaoL L, HuoC F, LiY W, et al. Structure and stability of the crystal Fe2C and low index surfaces[J]. Journal of Fuel Chemistry and Technology, 2009, 37: 104-108. |
| 15 | ChenW, LinT J, SunY H, et al. Recent advances in the investigation of nanoeffects of Fischer-Tropsch catalysts[J]. Catalysis Today, 2018, 311: 8-22. |
| 16 | PhamT H, QiY, YangJ, et al. Insights into Hägg iron-carbide-catalyzed Fischer-Tropsch synthesis: suppression of CH4 formation and enhancement of C-C coupling on χ-Fe5C2 (510)[J]. ACS Catalysis, 2015, 5(4): 2203-2208. |
| 17 | WangY, LiY, HuangS, et al. Insight into CH4 formation and chain growth mechanism of Fischer-Tropsch synthesis on θ-Fe3C (031)[J]. Chemical Physics Letters, 2017, 682: 115-121. |
| 18 | PerdewJ P, YueW. Accurate and simple density functional for the electronic exchange energy: generalized gradient approximation[J]. Physical Review B, 1986, 33(12): 8800. |
| 19 | PerdewJ P, BurkeK, ErnzerhofM. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865. |
| 20 | KresseG, JoubertD. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59(3): 1758. |
| 21 | HenkelmanG, JónssonH. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives[J]. The Journal of Chemical Physics, 1999, 111(15): 7010-7022. |
| 22 | HuoC F, LiY W, WangJ, et al. Insight into CH4 formation in iron-catalyzed Fischer-Tropsch synthesis[J]. Journal of the American Chemical Society, 2009, 131(41): 14713-14721. |
| 23 | 宋楠, 段学志, 周兴贵, 等. 费托催化剂η-Fe2C(011)上CO吸附与活化行为[J]. 陕西师范大学学报(自然科学版), 2019, 47(1): 13-19. |
| SongN, DuanX Z, ZhouX G, et al. Adsorption and activation of CO on η-Fe2C(011) surface of Fischer-Tropsch synthesis catalyst[J]. Journal of Shaanxi Normal University (Natural Science Edition), 2019, 47(1): 13-19. | |
| 24 | LoJ M H, ZieglerT. Density functional theory and kinetic studies of methanation on iron surface[J]. The Journal of Physical Chemistry C, 2007, 111(29): 11012-11025. |
| 25 | GovenderA, CurullaF D, NiemantsverdrietJ W. A density functional theory study on the effect of zero-point energy corrections on the methanation profile on Fe (100)[J]. ChemPhysChem, 2012, 13(6): 1591-1596. |
| 26 | ChengJ, HuP, EllisP, et al. Density functional theory study of iron and cobalt carbides for Fischer-Tropsch synthesis[J]. The Journal of Physical Chemistry C, 2010, 114(2): 1085-1093. |
| 27 | FischerF, TropschH. The preparation of synthetic oil mixtures (synthol) from carbon monoxide and hydrogen[J]. Brennstoff-Chem, 1923, 4: 276-285. |
| 28 | PichlerH, SchultzH. New insights in the area of the synthesis of hydrocarbons from CO and H2[J]. Chem. Ing. Tech., 1970, 12(18): 1160-1174. |
| 29 | KummerJ T, EmmettP H. Fischer-Tropsch synthesis mechanism studies. The addition of radioactive alcohols to the synthesis gas[J]. Journal of the American Chemical Society, 1953, 75(21): 5177-5183. |
| 30 | Ciobı̂căI M, KramerG J, GeQ, et al. Mechanisms for chain growth in Fischer-Tropsch synthesis over Ru (0001)[J]. Journal of Catalysis, 2002, 212(2): 136-144. |
| 31 | LiuZ P, HuP. A new insight into Fischer-Tropsch synthesis[J]. Journal of the American Chemical Society, 2002, 124(39): 11568-11569. |
| 32 | LoJ M H, ZieglerT. Theoretical studies of the formation and reactivity of C2 hydrocarbon species on the Fe (100) surface[J]. The Journal of Physical Chemistry C, 2007, 111(35): 13149-13162. |
| 33 | ZhaoY H, SunK, MaX, et al. Carbon chain growth by formyl insertion on rhodium and cobalt catalysts in syngas conversion[J]. Angewandte Chemie International Edition, 2011, 50(23): 5335-5338. |
| 34 | MichaelidesA, HuP. Insight into microscopic reaction pathways in heterogeneous catalysis[J]. Journal of the American Chemical Society, 2000, 122(40): 9866-9867. |
| 35 | ChengJ, GongX Q, HuP, et al. A quantitative determination of reaction mechanisms from density functional theory calculations: Fischer-Tropsch synthesis on flat and stepped cobalt surfaces[J]. Journal of Catalysis, 2008, 254(2): 285-295. |
| 36 | ChengJ, HuP, EllisP, et al. An energy descriptor to quantify methane selectivity in Fischer-Tropsch synthesis: a density functional theory study[J]. The Journal of Physical Chemistry C, 2009, 113(20): 8858-8863. |
| 37 | PeterM M, ValerioZ. The role of electrophilic species in the Fischer-Tropsch reaction[J]. Chemical Communications, 2009, 40(27): 1619-1634. |
| 38 | ChengJ, HuP, EllisP, et al. Chain growth mechanism in Fischer-Tropsch synthesis: a DFT study of C—C coupling over Ru, Fe, Rh, and Re surfaces[J]. The Journal of Physical Chemistry C, 2008, 112: 6082-6086. |
| 39 | CaoD B, LiY W, JiaoH J, et al. Chain growth mechanism of Fischer-Tropsch synthesis on Fe5C2 (001)[J]. Journal of Molecular Catalysis A: Chemical, 2011, 346: 55-69. |
| 40 | ZhaoY H, SunK, LiW X, et al. Carbon chain growth by formyl insertion on rhodium and cobalt catalysts in syngas conversion[J]. Angewandte Chemie International Edition, 2011, 50: 5335-5338. |
| 41 | SorescuD C. First-principles calculations of the adsorption and hydrogenation reactions of CHx (x= 0, 4) species on a Fe (100) surface[J]. Physical Review B, 2006, 73(15): 155420. |
| 42 | CaoD B, LiY W, WangJ, et al. Adsorption and reaction of surface carbon species on Fe5C2 (001)[J]. The Journal of Physical Chemistry C, 2008, 112(38): 14883-14890. |
| 43 | ParkJ Y, LeeY J, KhannaP K, et al. Alumina-supported iron oxide nanoparticles as Fischer-Tropsch catalysts: effect of particle size of iron oxide[J]. Journal of Molecular Catalysis A: Chemical, 2010, 323(1/2): 84-90. |
| 44 | TorresG H M, BitterJ H, DavidianT, et al. Iron particle size effects for direct production of lower olefins from synthesis gas[J]. Journal of the American Chemical Society, 2012, 134(39): 16207-16215. |
| 45 | GovenderA, Curulla-FerréD, Pérez-JigatoM, et al. First-principles elucidation of the surface chemistry of the C2Hx (x= 0—6) adsorbate series on Fe (100)[J]. Molecules, 2013, 18(4): 3806-3824. |
| [1] | Ke JIN, Chenguang WANG, Longlong MA, Qi ZHANG. Preparation of core-shell nanomaterials and their application in thermocatalytic hydrogenation of CO/CO2 [J]. CIESC Journal, 2022, 73(3): 990-1007. |
| [2] | TAN Khangwei, XIONG Wenting, FU Jile, CHEN Binghui. Preparation and catalytic performance of Ru-Co/SiC catalysts for the synthesis of heavy hydrocarbons from syngas by Fischer-Tropsch reaction [J]. CIESC Journal, 2021, 72(7): 3648-3657. |
| [3] | ZHANG Jianli, WANG Xu, MA Liping, YU Xufei, MA Qingxiang, FAN Subing, ZHAO Tiansheng. Preparation of modified MgFeMn-HTLcs and catalytic performance in CO hydrogenation [J]. CIESC Journal, 2018, 69(5): 2073-2080. |
| [4] | ZHANG Jun, ZHANG Zhengpai, SU Junjie, FU Donglong, DAI Weiwei, LIU Da, XU Jing, HAN Yifan. Effect of support basicity on iron-based catalysts for Fischer-Tropsch synthesis [J]. CIESC Journal, 2016, 67(2): 549-556. |
| [5] | LIU Yi, LIU Yong, CHEN Jianfeng, ZHANG Yi. Effects of MnOx supports on light olefin synthesis using cobalt catalyst in Fischer-Tropsch reaction [J]. CIESC Journal, 2015, 66(9): 3413-3420. |
| [6] | WANG Yan1,GE Xihui2,ZHANG Minqing1,ZHU Huaigong3,ZHANG Zijian2,WANG Ming1. Separation of n-hydrocarbons from high temperature oil phase products of Fischer-Tropsch synthesis [J]. Chemical Industry and Engineering Progree, 2014, 33(11): 2894-2898. |
| [7] | SUN Qiwen, WU Jianmin, ZHANG Zongsen, PANG Lifeng. Indirect coal liquefaction technology and its research progress [J]. Chemical Industry and Engineering Progree, 2013, 32(01): 1-12. |
| [8] | WANG Xiangyun. Progress of carbon dioxide removal from the recycle gas of F-T synthesis [J]. , 2011, 30(1): 52-. |
| [9] | JI Yuguo,ZHAO Zhen,YU Changchun,DUAN Aijun,JIANG Guiyuan. Progress of cobalt-based catalysts in Fischer-Tropsch synthesis [J]. , 2007, 26(7): 927-. |
| [10] | WANG Xiangyun. Progress of carbon dioxide removal from the recycle gas of Fischer-Tropsch synthesis [J]. , 2007, 26(12): 1708-. |
| [11] |
YANG Xiazhen,LIU Huazhang,TANG Haodong,CAI Liping,WU Zaiguo.
Research progress of promoters for Fe,Co-based Fischer-Tropsch synthesis catalysts [J]. , 2006, 25(8): 867-. |
| [12] |
DAI Xiaoping, YU Changchun, LI Qiang, ZHANG Changbin, JIANG Qiying, SHEN Shikong.
An Integrated Process of a Two-Stage Fixed Bed Syngas Production and F-T Synthesis for GTL in Remote Gas Field [J]. , 2003, 11(1): 85-89. |
| [13] |
LU Zhaohui, ZHAO Yulong, ZHAO Liangfu.
Two-Bubble Class Model for Churn-Turbulent Regime in a Bubble Column Slurry Reactor of Fischer-Tropsch Synthesis [J]. , 2001, 9(1): 84-89. |
| [14] | Ding Baiquan, Li Tao, A.A.C.M. Beenackers, C.P.van der laan. Overview of Fischer-Tropsch Synthesis in Slurry Reactors [J]. , 2000, 8(3): 255-266. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

