CIESC Journal ›› 2022, Vol. 73 ›› Issue (9): 3828-3840.DOI: 10.11949/0438-1157.20220575
• Reviews and monographs • Previous Articles Next Articles
Songtao YANG1( ), Dongyang LI2(), Yuqing NIU4, Xingang LI1, Shaohui KANG4, Hong LI1, Kaikai YE4, Zhiquan ZHOU4, Xin GAO1,3()
), Dongyang LI2(), Yuqing NIU4, Xingang LI1, Shaohui KANG4, Hong LI1, Kaikai YE4, Zhiquan ZHOU4, Xin GAO1,3()
Received:2022-04-24
Revised:2022-07-01
Online:2022-10-09
Published:2022-09-05
Contact:
Dongyang LI, Xin GAO
杨松涛1(), 李东洋2(), 牛玉清4, 李鑫钢1, 康绍辉4, 李洪1, 叶开凯4, 周志全4, 高鑫1,3()
通讯作者:
李东洋,高鑫
作者简介:杨松涛(1997—),男,硕士研究生,yangsongtao@tju.edu.cn
基金资助:CLC Number:
Songtao YANG, Dongyang LI, Yuqing NIU, Xingang LI, Shaohui KANG, Hong LI, Kaikai YE, Zhiquan ZHOU, Xin GAO. Molecular simulation progress in studying thermodynamic properties and potential functions of fluorides[J]. CIESC Journal, 2022, 73(9): 3828-3840.
杨松涛, 李东洋, 牛玉清, 李鑫钢, 康绍辉, 李洪, 叶开凯, 周志全, 高鑫. 氟化物势能函数和热力学性质的分子模拟研究进展[J]. 化工学报, 2022, 73(9): 3828-3840.
Add to citation manager EndNote|Ris|BibTeX

Fig.1 Molecular configuration, physical properties and industrial applications of fluorides
| 分类 | 方法 | 特点 |
|---|---|---|
| 直接模拟法 | GEMC | 适合简单流体和较复杂流体 |
| CBMC | 适合复杂大分子或含有氢键等强相互作用的稠密流体 | |
| RGEMC | 适合模拟反应体系的相平衡 | |
| 间接模拟法 | NPT+TP | 模拟时间长,较为烦琐,应用较少 |
| GCMC | 适合非均相系统 | |
| GDI | 主要用于纯物质的计算,应用较多 | |
| HRW | 气液相波动剧烈的临界区的模拟 | |
| 新方法 | HRW和GEMC相结合 | 尚不成熟 |
| kMC | 适合模拟稀薄流体和缔合流体的相平衡 | |
| Bin-CMC | 气固相平衡 |
Table 1 Classification and characteristics of Monte Carlo method
| 分类 | 方法 | 特点 |
|---|---|---|
| 直接模拟法 | GEMC | 适合简单流体和较复杂流体 |
| CBMC | 适合复杂大分子或含有氢键等强相互作用的稠密流体 | |
| RGEMC | 适合模拟反应体系的相平衡 | |
| 间接模拟法 | NPT+TP | 模拟时间长,较为烦琐,应用较少 |
| GCMC | 适合非均相系统 | |
| GDI | 主要用于纯物质的计算,应用较多 | |
| HRW | 气液相波动剧烈的临界区的模拟 | |
| 新方法 | HRW和GEMC相结合 | 尚不成熟 |
| kMC | 适合模拟稀薄流体和缔合流体的相平衡 | |
| Bin-CMC | 气固相平衡 |
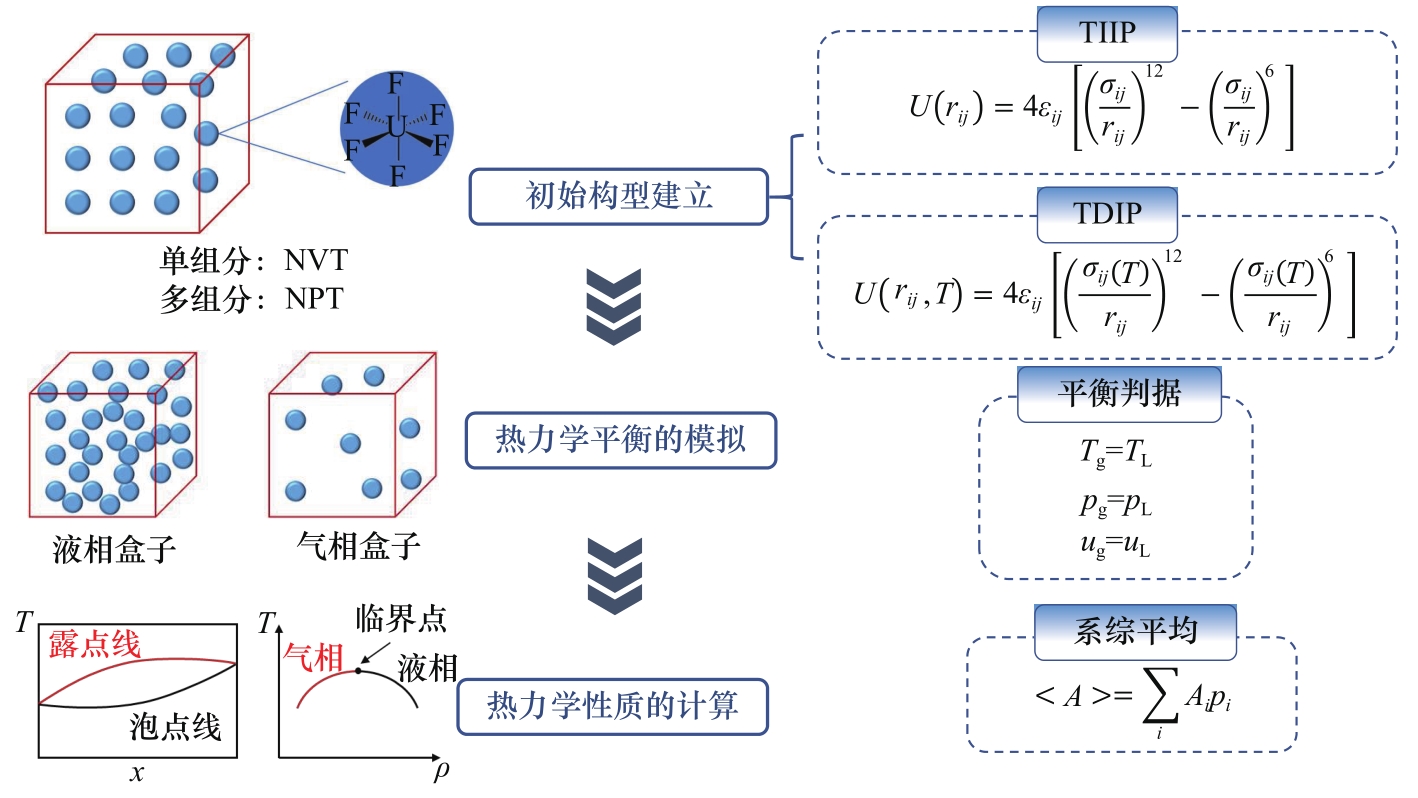
Fig.2 GEMC method for thermodynamic properties of fluoride phase equilibrium

Fig.3 Lennard-Jones (n, 6) potential curves
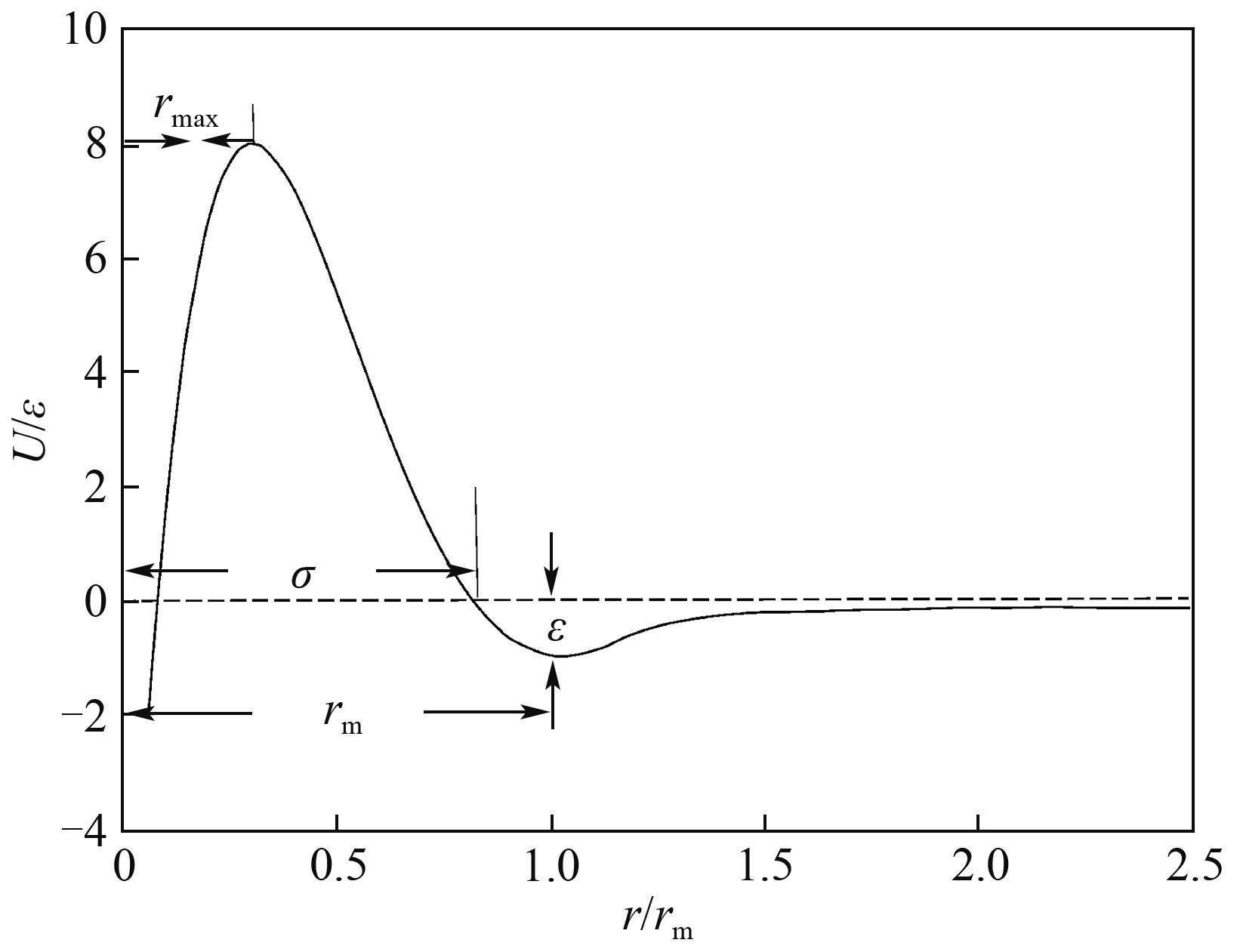
Fig.4 Modified Buckingham (exp-6) potential curves

Fig.5 Temperature dependence of rm(T) and ε(T) of UF6
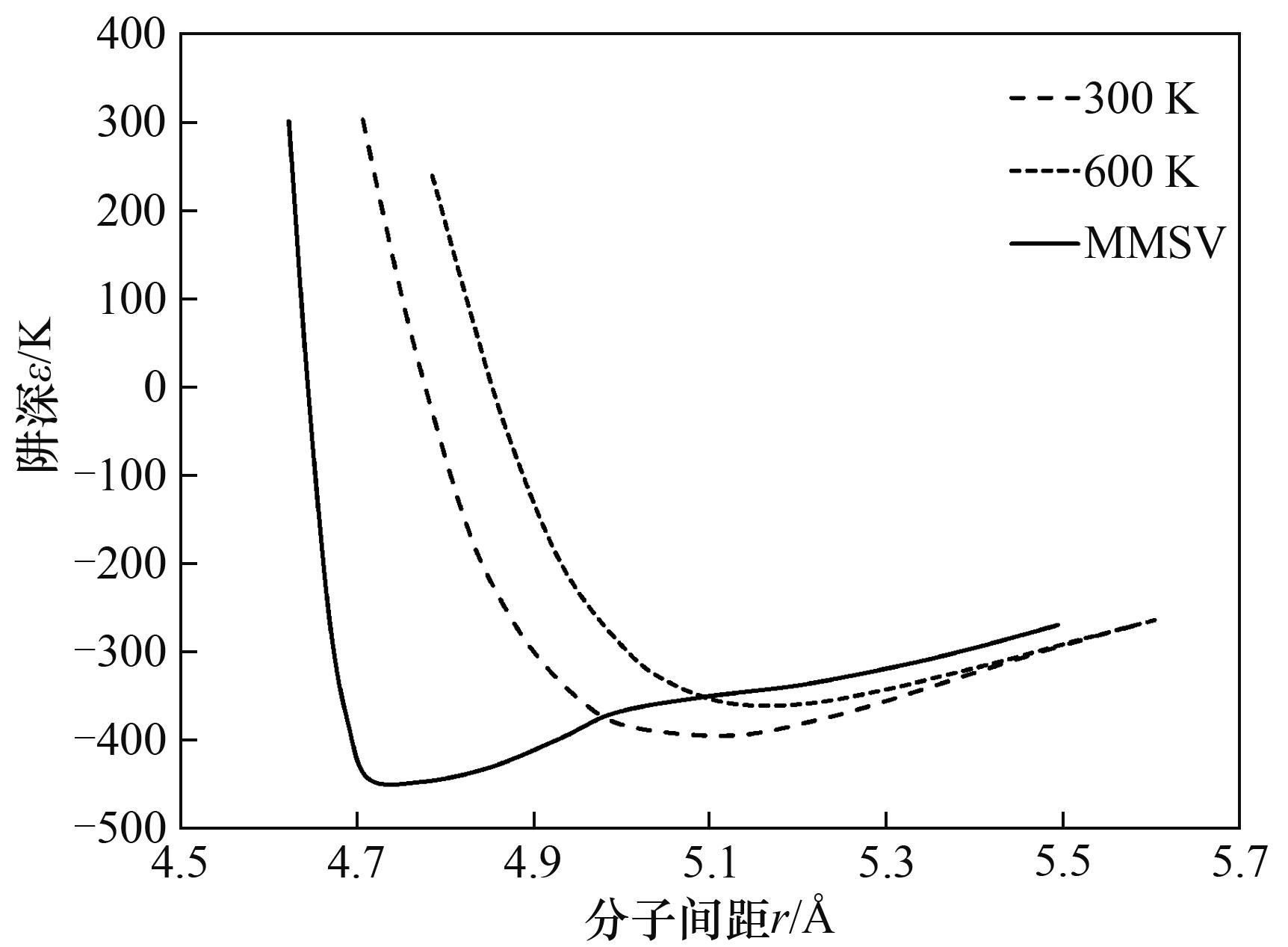
Fig.6 Comparison of potential curves between TDIP and MMSV(SF6) [15]
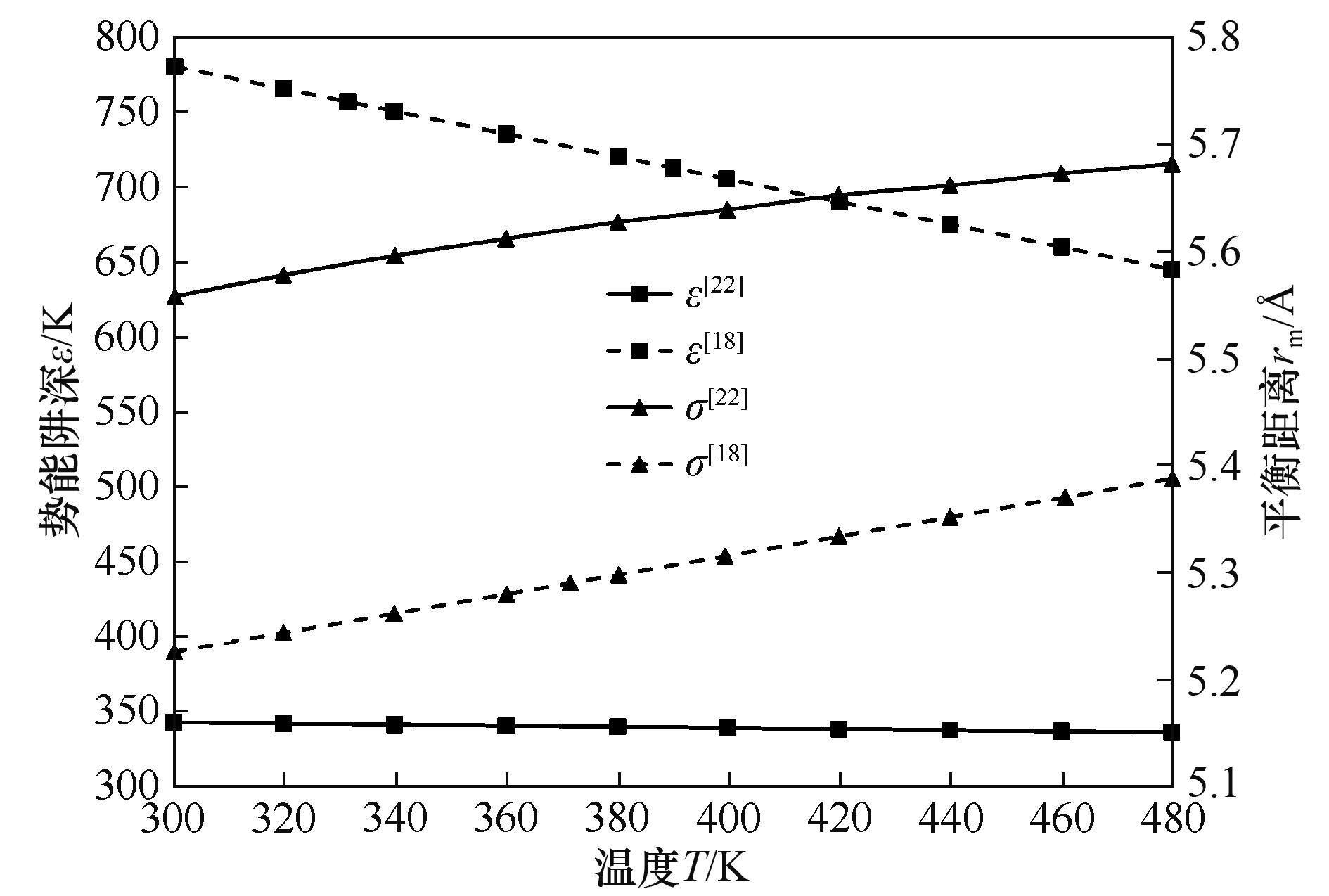
Fig.7 Effect of temperature on UF6 collision diameter (σ) and well depth (ε)
| 方法 | 文献 | RMSD(B) |
|---|---|---|
| TDIP | [ | 35.449 |
| [ | 3.8931 | |
| TIIP | [ | 38.895 |
| [ | 151.31 | |
| [ | 147.66 | |
| [ | 261.01 | |
| [ | 11.187 |
Table 2 RMSD of second virial coefficient of UF6 in different literatures
| 方法 | 文献 | RMSD(B) |
|---|---|---|
| TDIP | [ | 35.449 |
| [ | 3.8931 | |
| TIIP | [ | 38.895 |
| [ | 151.31 | |
| [ | 147.66 | |
| [ | 261.01 | |
| [ | 11.187 |

Fig.8 Vapor-liquid coexistence curves (TDIP and TIIP) and experimental data of UF6
| 数据来源 | RMSD (B)/(cm3/mol) | |||
|---|---|---|---|---|
| Oh关联式 | Tsonopoulos 关联式 | Dymond关联式 | Zarkova & Hohm关联式 | |
| Ref.[ | 71 | 49.7 | 98.9 | 70.8 |
| Ref.[ | 77 | 94.3 | 40 | 77.3 |
| all data | 68 | 82.6 | 65.1 | 75.2 |
| 数据来源 | RMSD (η)/% | |||
| Oh关联式 | Lucas关联式 | Zarkova & Hohm关联式 | ||
| Ref.[ | 3.2 | 3 | 1.8 | |
| Ref.[ | 2.9 | 4.2 | 0.7 | |
| Ref.[ | 3.7 | 4.9 | 2.3 | |
| Ref.[ | 3.5 | 4.8 | 0.7 | |
| all data | 3.4 | 4.4 | 1.5 | |
Table 3 RMSD of calculation correlation and experimental data of second virial coefficient and viscosity of UF6
| 数据来源 | RMSD (B)/(cm3/mol) | |||
|---|---|---|---|---|
| Oh关联式 | Tsonopoulos 关联式 | Dymond关联式 | Zarkova & Hohm关联式 | |
| Ref.[ | 71 | 49.7 | 98.9 | 70.8 |
| Ref.[ | 77 | 94.3 | 40 | 77.3 |
| all data | 68 | 82.6 | 65.1 | 75.2 |
| 数据来源 | RMSD (η)/% | |||
| Oh关联式 | Lucas关联式 | Zarkova & Hohm关联式 | ||
| Ref.[ | 3.2 | 3 | 1.8 | |
| Ref.[ | 2.9 | 4.2 | 0.7 | |
| Ref.[ | 3.7 | 4.9 | 2.3 | |
| Ref.[ | 3.5 | 4.8 | 0.7 | |
| all data | 3.4 | 4.4 | 1.5 | |
| 1 | 邓茗文. 中国核电:硬核助力“双碳”目标 清洁赋能美好未来[J]. 可持续发展经济导刊, 2021(8): 44-48. |
| Deng M W. CNNP: developing the nuclear power to achieve carbon emission peaking and carbon-neutral target of China[J]. China Sustainability Tribune, 2021(8): 44-48. | |
| 2 | 吕程, 鞠吉, 曾爱武. 苯-噻吩-NMP三元体系汽液平衡的GEMC模拟[J]. 计算机与应用化学, 2016, 33(3): 309-312. |
| Lv C, Ju J, Zeng A W. Gemc simulation for benzene/thiophene/nmp ternary system[J]. Computers and Applied Chemistry, 2016, 33(3): 309-312. | |
| 3 | 吴东, 祁影霞, 杨喜. HFO-1234YF/HFC-32气-液相平衡特性研究[J]. 制冷技术, 2016, 36(1): 26-30, 34. |
| Wu D, Qi Y X, Yang X. Investigation on vapor-liquid equilibrium characteristics of HFO-1234YF/HFC-32[J]. Chinese Journal of Refrigeration Technology, 2016, 36(1): 26-30, 34. | |
| 4 | 董秀芹, 管肖肖, 马静. 二氧化碳-醋酸乙烯体系相平衡的GEMC模拟[J]. 计算机与应用化学, 2015, 32(10): 1182-1186. |
| Dong X Q, Guan X X, Ma J. Gibbs ensemble Monte Carlo simulations of binary mixture of CO2 and vinyl acetate[J]. Computers and Applied Chemistry, 2015, 32(10): 1182-1186. | |
| 5 | Potoff J J, Siepmann J I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen[J]. AIChE Journal, 2001, 47(7): 1676-1682. |
| 6 | Li H, Zhang J, Li D Y, et al. Monte Carlo simulations of vapour-liquid phase equilibrium and microstructure for the system containing azeotropes[J]. Molecular Simulation, 2017, 43(13/14/15/16): 1125-1133. |
| 7 | Li D Y, Gao Z Q, Vasudevan N K, et al. Molecular mechanism for azeotrope formation in ethanol/benzene binary mixtures through Gibbs ensemble Monte Carlo simulation[J]. The Journal of Physical Chemistry. B, 2020, 124(16): 3371-3386. |
| 8 | Malyshev V V. Equation of state for UF6 over the range of density variation to 0.0118 g/cm3 and temperature variation up to 367 K[J]. Atomic Energy (USSR), 1973, 34(1): 42-44. |
| 9 | Malyshev V V. Equations of state for UF6 for a wide range of state parameters[J]. Atomic Energy (USSR), 32(4):313-314. |
| 10 | Morizot P, Ostorero J, Plurien P. Viscosité et non-idéalité des hexafluorures de molybdène, de tungstène, d’uranium détermination de leurs paramètres moléculaires[J]. Journal De Chimie Physique, 1973, 70: 1582-1586. |
| 11 | Schneider B, Boring A M, Cohen J S. Interaction potentials for UF6 with itself and with rare-gas atoms[J]. Chemical Physics Letters, 1974, 27(4): 577-579. |
| 12 | Heintz A, Meisinger E, Lichtenthaler R N. Measured values of the second virial coefficient and determining the intermolecular interaction potential of gaseous uranium hexafluoride[J]. Berichte der Bunsengesellschaft fuer Physikalische Chemie, 1976, 80(2): 163-166. |
| 13 | Heintz A, Lichtenthaler R N. Data of the second virial coefficient of the gases PF5, MoF6, WF6 and JF5 and the determination of the intermolecular interaction potential[J]. Berichte Der Bunsengesellschaft/Physical Chemistry Chemical Physics, 1976, 80(10): 962-965. |
| 14 | Kirch P, Schütte R. Measurement of thermal diffusion and determination of the intermolecular potential of gaseous uranium hexafluoride[J]. The Journal of Chemical Physics, 1965, 42(10): 3729-3730. |
| 15 | Aziz R A, Slaman M J, Taylor W L, et al. An improved intermolecular potential for sulfur hexafluoride[J]. The Journal of Chemical Physics, 1991, 94(2): 1034-1038. |
| 16 | Coroiu I, Demco D E. Second virial coefficients and transport properties of hexafluoride gases from an improved intermolecular potential[J]. Zeitschrift Für Naturforschung A, 1997, 52(10): 748-756. |
| 17 | El-Kader M S A, Kalugina Y N. Dipole-octupole polarisability of uranium hexafluoride and theoretical prediction of anisotropic light-scattering spectrum using new intermolecular potential[J]. Molecular Physics, 2016, 114(1): 44-52. |
| 18 | Zarkova L, Pirgov P. Transport and equilibrium properties of UF6 gas simultaneously fitted by an effective isotropic potential with temperature-dependent parameters[J]. Journal of Physics B: Atomic, Molecular and Optical Physics, 1995, 28(19): 4269-4281. |
| 19 | Zarkova L. An isotropic intermolecular potential with temperature dependent effective parameters for heavy globular gases[J]. Molecular Physics, 1996, 88(2): 489-495. |
| 20 | Zarkova L, Hohm U. pVT-second virial coefficients B(T ), viscosity η(T ), and self-diffusion ρD(T) of the gases: BF3, CF4, SiF4, CCl4, SiCl4, SF6, MoF6, WF6, UF6, C(CH3)4, and Si(CH3)4 determined by means of an isotropic temperature-dependent potential[J]. Journal of Physical and Chemical Reference Data, 2002, 31(1): 183-216. |
| 21 | Zarkova L, Hohm U, Damyanova M. Viscosity and pVT-second virial coefficient of binary noble-globular gas and globular-globular gas mixtures calculated by means of an isotropic temperature-dependent potential[J]. Journal of Physical and Chemical Reference Data, 2003, 32(4): 1591-1705. |
| 22 | Al-Matar A K, Binous H. Vapor-liquid phase equilibrium diagram for uranium hexafluoride (UF6) using simplified temperature dependent intermolecular potential parameters (TDIP)[J]. Journal of Radioanalytical and Nuclear Chemistry, 2016, 310(1): 139-154. |
| 23 | Oh S K. Application of the group contribution concept to Kihara potential for estimating thermodynamic and transport properties(Ⅵ): Heavy globular molecules (SF6, MoF6, WF6, UF6, C(CH3)4, Si(CH3)4)[J]. Fluid Phase Equilibria, 2008, 271(1/2): 53-68. |
| 24 | Sadus R J. Molecular Simulation of Fluids[M]. Amsterdam:Elsevier, 2002. |
| 25 | Dove M T, Pawley G S. A molecular dynamics simulation study of the plastic crystalline phase of sulphur hexafluoride[J]. Journal of Physics C: Solid State Physics, 1983, 16(31): 5969-5983. |
| 26 | Lu H. Molecular dynamics simulations of solid sulphur hexafluoride[D]. Edinburgh: The University of Edinburgh, 1992. |
| 27 | 李洪, 张季, 李鑫钢, 等. 分子模拟方法计算相平衡热力学性质的研究进展[J]. 化工进展, 2017, 36(8): 2731-2741. |
| Li H, Zhang J, Li X G, et al. Progress in study on thermodynamic properties of phase equilibria using molecular simulation[J]. Chemical Industry and Engineering Progress, 2017, 36(8): 2731-2741. | |
| 28 | Rosenbluth M N, Rosenbluth A W. Monte Carlo calculation of the average extension of molecular chains[J]. The Journal of Chemical Physics, 1955, 23(2): 356-359. |
| 29 | Lísal M, Smith W R, Nezbeda I. Accurate vapour-liquid equilibrium calculations for complex systems using the reaction Gibbs ensemble Monte Carlo simulation method[J]. Fluid Phase Equilibria, 2001, 181(1/2): 127-146. |
| 30 | Lotfi A, Vrabec J, Fischer J. Vapour liquid equilibria of the Lennard-Jones fluid from the NpT plus test particle method[J]. Molecular Physics, 1992, 76(6): 1319-1333. |
| 31 | Yao J, Greenkorn R A, Chao K C. Monte Carlo simulation of the grand canonical ensemble[J]. Molecular Physics, 1982, 46(3): 587-594. |
| 32 | Kofke D A. Gibbs-Duhem integration: a new method for direct evaluation of phase coexistence by molecular simulation[J]. Molecular Physics, 1993, 78(6): 1331-1336. |
| 33 | Potoff J J, Panagiotopoulos A Z. Surface tension of the three-dimensional Lennard-Jones fluid from histogram-reweighting Monte Carlo simulations[J]. The Journal of Chemical Physics, 2000, 112(14): 6411-6415. |
| 34 | Boulougouris G C, Peristeras L D, Economou I G, et al. Predicting fluid phase equilibrium via histogram reweighting with Gibbs ensemble Monte Carlo simulations[J]. The Journal of Supercritical Fluids, 2010, 55(2): 503-509. |
| 35 | Ustinov E A, Do D D. Application of kinetic Monte Carlo method to equilibrium systems: vapour-liquid equilibria[J]. Journal of Colloid and Interface Science, 2012, 366(1): 216-223. |
| 36 | Nguyen V T, Tan S J, Do D D, et al. Application of kinetic Monte Carlo method to the vapour-liquid equilibria of associating fluids and their mixtures[J]. Molecular Simulation, 2016, 42(8): 642-654. |
| 37 | Fan C Y, Do D D, Nicholson D. New Monte Carlo simulation of adsorption of gases on surfaces and in pores: a concept of multibins[J]. The Journal of Physical Chemistry. B, 2011, 115(35): 10509-10517. |
| 38 | Fan C Y, Do D D, Nicholson D. A new and effective Bin-Monte Carlo scheme to study vapour-liquid equilibria and vapour-solid equilibria[J]. Fluid Phase Equilibria, 2012, 325: 53-65. |
| 39 | Jorgensen W L, Maxwell D S, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids[J]. Journal of the American Chemical Society, 1996, 118(45): 11225-11236. |
| 40 | Sun H. COMPASS: an ab initio force-field optimized for condensed-phase. Applications overview with details on alkane and benzene compounds[J]. The Journal of Physical Chemistry B, 1998, 102(38): 7338-7364. |
| 41 | Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields[J]. Journal of Computational Chemistry, 2010, 31(4): 671-690. |
| 42 | Martin M G, Siepmann J I. Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes[J]. The Journal of Physical Chemistry B, 1999, 103(21): 4508-4517. |
| 43 | Martin M G, Siepmann J I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes[J]. The Journal of Physical Chemistry B, 1998, 102(14): 2569-2577. |
| 44 | Eggimann B L, Sunnarborg A J, Stern H D, et al. An online parameter and property database for the TraPPE force field[J]. Molecular Simulation, 2014, 40(1/2/3): 101-105. |
| 45 | Nath S K, Escobedo F A, de Pablo J J, et al. Simulation of vapor-liquid equilibria for alkane mixtures[J]. Industrial & Engineering Chemistry Research, 1998, 37(8): 3195-3202. |
| 46 | Marrink S J, de Vries A H, Mark A E. Coarse grained model for semiquantitative lipid simulations[J]. The Journal of Physical Chemistry B, 2004, 108(2): 750-760. |
| 47 | Shinoda W, DeVane R, Klein M L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants[J]. Molecular Simulation, 2007, 33(1/2): 27-36. |
| 48 | Olivet A, Vega L F. Optimized molecular force field for sulfur hexafluoride simulations[J]. The Journal of Chemical Physics, 2007, 126(14): 144502. |
| 49 | Lee K H, Lombardo M, Sandler S I. The generalized van der Waals partition function(Ⅱ): Application to the square-well fluid[J]. Fluid Phase Equilibria, 1985, 21(3): 177-196. |
| 50 | Stone A. The Theory of Intermolecular Forces[M]. Oxford: Oxford University Press, 2013. |
| 51 | Rice W E, Hirschfelder J O. Second virial coefficients of gases obeying a modified Buckingham (exp-six) potential[J]. The Journal of Chemical Physics, 1954, 22(2): 187-192. |
| 52 | Kihara T. Intermolecular Forces[M]. New York: John Wiley & Sons, 1978. |
| 53 | Parson J M, Siska P E, Lee Y T. Intermolecular potentials from crossed-beam differential elastic scattering measurements (Ⅳ): Ar+Ar[J]. The Journal of Chemical Physics, 1972, 56(4): 1511-1516. |
| 54 | Pack R T, Piper E, Pfeffer G A, et al. Multiproperty empirical anisotropic intermolecular potentials (Ⅱ): HeSF6 and NeSF6 [J]. The Journal of Chemical Physics, 1984, 80(10): 4940-4950. |
| 55 | Pack R T, Valentini J J, Becker C H, et al. Multiproperty empirical interatomic potentials for ArXe and KrXe[J]. The Journal of Chemical Physics, 1982, 77(11): 5475-5485. |
| 56 | Meng L, Duan Y Y. Site-site potential function and second virial coefficients for linear molecules[J]. Molecular Physics, 2006, 104(18): 2891-2899. |
| 57 | Zhang Y, Wang S, He M G. Modified 2CLJDQP model and the second virial coefficients for linear molecules[J]. Chinese Physics B, 2014, 23(12): 125101. |
| 58 | Meinander N. An isotropic intermolecular potential for sulfur hexafluoride based on the collision-induced light scattering spectrum, viscosity, and virial coefficient data[J]. The Journal of Chemical Physics, 1993, 99(11): 8654-8667. |
| 59 | Stefanov B, Zarkova L. The equilibrium and transport properties of heavy fluorine-containing gases predicted with the use of the vibrationally-excited-states-of-molecules (VESM) model[J]. High Temperatures-High Pressures, 1993, 25: 481-486. |
| 60 | Zarkova L, Pirgov P. The isotropic temperature-dependent potential describing the binary interactions in gaseous and[J]. Journal of Physics B: Atomic, Molecular and Optical Physics, 1996, 29(19): 4411-4422. |
| 61 | Stefanov B, Zarkova L. The model of vibrationally excited states of molecules as a tool for calculating thermodynamic and transport properties of molecular gases: SF6 as an example[J]. High Temperatures-High Pressures, 1993, 25: 487-490. |
| 62 | Zarkova L, Pirgov P. Thermophysical properties of diluted F-containing heavy globular gases predicted by means of temperature dependent effective isotropic potential[J]. Vacuum, 1997, 48(1): 21-27. |
| 63 | Zarkova L, Hohm U. Effective (n-6) Lennard-Jones potentials with temperature-dependent parameters introduced for accurate calculation of equilibrium and transport properties of ethene, propene, butene, and cyclopropane[J]. Journal of Chemical & Engineering Data, 2009, 54(6): 1648-1655. |
| 64 | Dymond J H, Marsh K N, Wilhoit R C, et al. Virial Coefficients of Pure Gases[M]. Landord-Bornstein: Springer, 2003. |
| 65 | DeWitt R. Uranium Hexafluoride: A Survey of The Physicochemical Properties[R]. Office of Scientific and Technical Information (OSTI), 1960. |
| 66 | Verkhivker G P, Tetel’baum S D, Konyaeva G P. Thermodynamic properties of uranium hexafluoride (UF6)[J]. Soviet Atomic Energy, 1968, 24(2): 191-195. |
| 67 | Malyshev V V. Kihara interaction potentials for octahedral molecular systems SF6, MoF6, WF6, and UF6 [J]. High Temperature, 1974, 12(5): 979-983. |
| 68 | Hellemans J M, Kestin J, Ro S T. The viscosity of CH4, CF4 and SF6 over a range of temperatures[J]. Physica, 1973, 65(2): 376-380. |
| 69 | Kestin J, Khalifa H E, Ro S T, et al. The viscosity and diffusion coefficients of eighteen binary gaseous systems[J]. Physica A: Statistical Mechanics and Its Applications, 1977, 88(2): 242-260. |
| 70 | Strehlow T, Vogel E. Temperature dependence and initial density dependence of the viscosity of sulphur hexafluoride[J]. Physica A: Statistical Mechanics and Its Applications, 1989, 161(1): 101-117. |
| 71 | Kestin J, Ro S T, Wakeham W A. Reference values of the viscosity of twelve gases at 25℃[J]. Trans. Faraday Soc., 1971, 67: 2308-2313. |
| 72 | Kestin J, Khalifa H E, Wakeham W A. Viscosity of multicomponent mixtures of four complex gases[J]. The Journal of Chemical Physics, 1976, 65(12): 5186-5188. |
| 73 | Kestin J, Khalifa H E, Wakeham W A. The viscosity of gaseous mixtures containing krypton[J]. The Journal of Chemical Physics, 1977, 67(9): 4254-4259. |
| 74 | Abe Y, Kestin J, Khalifa H E, et al. The viscosity and diffusion coefficients of the mixtures of light hydrocarbons with other polyatomic gases[J]. Berichte Der Bunsengesellschaft Für Physikalische Chemie, 1979, 83(3): 271-276. |
| 75 | Hoogland J H B, van den Berg H R, Trappeniers N J. Measurements of the viscosity of sulfur hexaflouride up to 100 bar by a capillary-flow viscometer[J]. Physica A: Statistical Mechanics and Its Applications, 1985, 134(1): 169-192. |
| 76 | Boushehri A, Bzowski J, Kestin J, et al. Equilibrium and transport properties of eleven polyatomic gases at low density[J]. Journal of Physical and Chemical Reference Data, 1987, 16(3): 445-466. |
| 77 | Trengove R D, Wakeham W A. The viscosity of carbon dioxide, methane, and sulfur hexafluoride in the limit of zero density[J]. Journal of Physical and Chemical Reference Data, 1987, 16(2): 175-187. |
| 78 | Lichtenthaler R N, Schafer K. Intermolecular forces of spherical and non-spherical molecules calculated from second virial coefficients[J]. Berichte der Bunsen-Gesellschaft fur Physikalische Chemie, 1969, 73(1): 42-+. |
| 79 | Bzowski J, Kestin J, Mason E A, et al. Equilibrium and transport properties of gas mixtures at low density: eleven polyatomic gases and five noble gases[J]. Journal of Physical and Chemical Reference Data, 1990, 19(5): 1179-1232. |
| 80 | Zarkova L, Hohm U, Damyanova M. Comparison of Lorentz-Berthelot and Tang-Toennies mixing rules using an isotropic temperature-dependent potential applied to the thermophysical properties of binary gas mixtures of CH4, CF4, SF6, and C(CH3)4 with Ar, Kr, and Xe[J]. International Journal of Thermophysics, 2004, 25(6): 1775-1798. |
| 81 | Tsonopoulos C. An empirical correlation of second virial coefficients[J]. AIChE Journal, 1974, 20(2): 263-272. |
| 82 | Lucas K. Phase Equilibria and Fluid Properties in the Chemical Industry[M]. Frankfurt: Dechema, 1980: 100. |
| 83 | Kigoshi K. On the viscosity of the uranium hexafluoride[J]. Bulletin of the Chemical Society of Japan, 1950, 23(2): 67-68. |
| 84 | Myerson A L, Eicher J H. The viscosity of gaseous uranium Hexafluoride1[J]. Journal of the American Chemical Society, 1952, 74(11): 2758-2761. |
| 85 | Llewellyn D R. 5. Some physical properties of uranium hexafluoride[J]. Journal of the Chemical Society (Resumed), 1953: 28. |
| [1] | Minghao SONG, Fei ZHAO, Shuqing LIU, Guoxuan LI, Sheng YANG, Zhigang LEI. Multi-scale simulation and study of volatile phenols removal from simulated oil by ionic liquids [J]. CIESC Journal, 2023, 74(9): 3654-3664. |
| [2] | Jianbo HU, Hongchao LIU, Qi HU, Meiying HUANG, Xianyu SONG, Shuangliang ZHAO. Molecular dynamics simulation insight into translocation behavior of organic cage across the cellular membrane [J]. CIESC Journal, 2023, 74(9): 3756-3765. |
| [3] | Jiajia ZHAO, Shixiang TIAN, Peng LI, Honggao XIE. Microscopic mechanism of SiO2-H2O nanofluids to enhance the wettability of coal dust [J]. CIESC Journal, 2023, 74(9): 3931-3945. |
| [4] | Linzheng WANG, Yubing LU, Ruizhi ZHANG, Yonghao LUO. Analysis on thermal oxidation characteristics of VOCs based on molecular dynamics simulation [J]. CIESC Journal, 2023, 74(8): 3242-3255. |
| [5] | Ji CHEN, Ze HONG, Zhao LEI, Qiang LING, Zhigang ZHAO, Chenhui PENG, Ping CUI. Study on coke dissolution loss reaction and its mechanism based on molecular dynamics simulations [J]. CIESC Journal, 2023, 74(7): 2935-2946. |
| [6] | Ming DONG, Jinliang XU, Guanglin LIU. Molecular dynamics study on heterogeneous characteristics of supercritical water [J]. CIESC Journal, 2023, 74(7): 2836-2847. |
| [7] | Yuanchao LIU, Xuhao JIANG, Ke SHAO, Yifan XU, Jianbin ZHONG, Zhuan LI. Influence of geometrical dimensions and defects on the thermal transport properties of graphyne nanoribbons [J]. CIESC Journal, 2023, 74(6): 2708-2716. |
| [8] | Xiaoyu YAO, Jun SHEN, Jian LI, Zhenxing LI, Huifang KANG, Bo TANG, Xueqiang DONG, Maoqiong GONG. Research progress in measurement methods in vapor-liquid critical properties of mixtures [J]. CIESC Journal, 2023, 74(5): 1847-1861. |
| [9] | Ke CHEN, Li DU, Ying ZENG, Siying REN, Xudong YU. Phase equilibria and calculation of quaternary system LiCl+MgCl2+CaCl2+H2O at 323.2 K [J]. CIESC Journal, 2023, 74(5): 1896-1903. |
| [10] | Hao GU, Fujian ZHANG, Zhen LIU, Wenxuan ZHOU, Peng ZHANG, Zhongqiang ZHANG. Desalination performance and mechanism of porous graphene membrane in temporal dimension under mechanical-electrical coupling [J]. CIESC Journal, 2023, 74(5): 2067-2074. |
| [11] | Chenxin LI, Yanqiu PAN, Liu HE, Yabin NIU, Lu YU. Carbon membrane model based on carbon microcrystal structure and its gas separation simulation [J]. CIESC Journal, 2023, 74(5): 2057-2066. |
| [12] | Yuanjing MAO, Zhi YANG, Songping MO, Hao GUO, Ying CHEN, Xianglong LUO, Jianyong CHEN, Yingzong LIANG. Estimation of SAFT-VR Mie equation of state parameters and thermodynamic properties of C6—C10 alcohols [J]. CIESC Journal, 2023, 74(3): 1033-1041. |
| [13] | Wenting CHENG, Jie LI, Li XU, Fangqin CHENG, Guoji LIU. Experiment and prediction for the solubility of AlCl3·6H2O in FeCl3, CaCl2, KCl and KCl-FeCl3 solutions [J]. CIESC Journal, 2023, 74(2): 642-652. |
| [14] | Yi LIAO, Yabin NIU, Yanqiu PAN, Lu YU. Modeling the effects of mixed surfactants on the behaviors and properties of the oil-water interface with molecular dynamics [J]. CIESC Journal, 2022, 73(9): 4003-4014. |
| [15] | Mo ZHENG, Xiaoxia LI. Revealing reaction compromise in competition for volatile radicals during coal pryolysis via ReaxFF MD simulation [J]. CIESC Journal, 2022, 73(6): 2732-2741. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

