CIESC Journal ›› 2025, Vol. 76 ›› Issue (3): 1084-1092.DOI: 10.11949/0438-1157.20240849
• Process system engineering • Previous Articles Next Articles
Xinyu ZHENG( ), Zehua REN, Li ZHOU, Shiyang CHAI(), Xu JI
), Zehua REN, Li ZHOU, Shiyang CHAI(), Xu JI
Received:2024-07-26
Revised:2024-09-18
Online:2025-03-28
Published:2025-03-25
Contact:
Shiyang CHAI
郑欣雨(), 任泽华, 周利, 柴士阳(), 吉旭
通讯作者:
柴士阳
作者简介:郑欣雨(2001—),女,硕士研究生,xinyuzheng_sc@163.com
基金资助:CLC Number:
Xinyu ZHENG, Zehua REN, Li ZHOU, Shiyang CHAI, Xu JI. Lattice energy regression model based on crystal graph convolutional neural networks[J]. CIESC Journal, 2025, 76(3): 1084-1092.
郑欣雨, 任泽华, 周利, 柴士阳, 吉旭. 基于晶体图卷积神经网络的晶格能回归模型[J]. 化工学报, 2025, 76(3): 1084-1092.
Add to citation manager EndNote|Ris|BibTeX
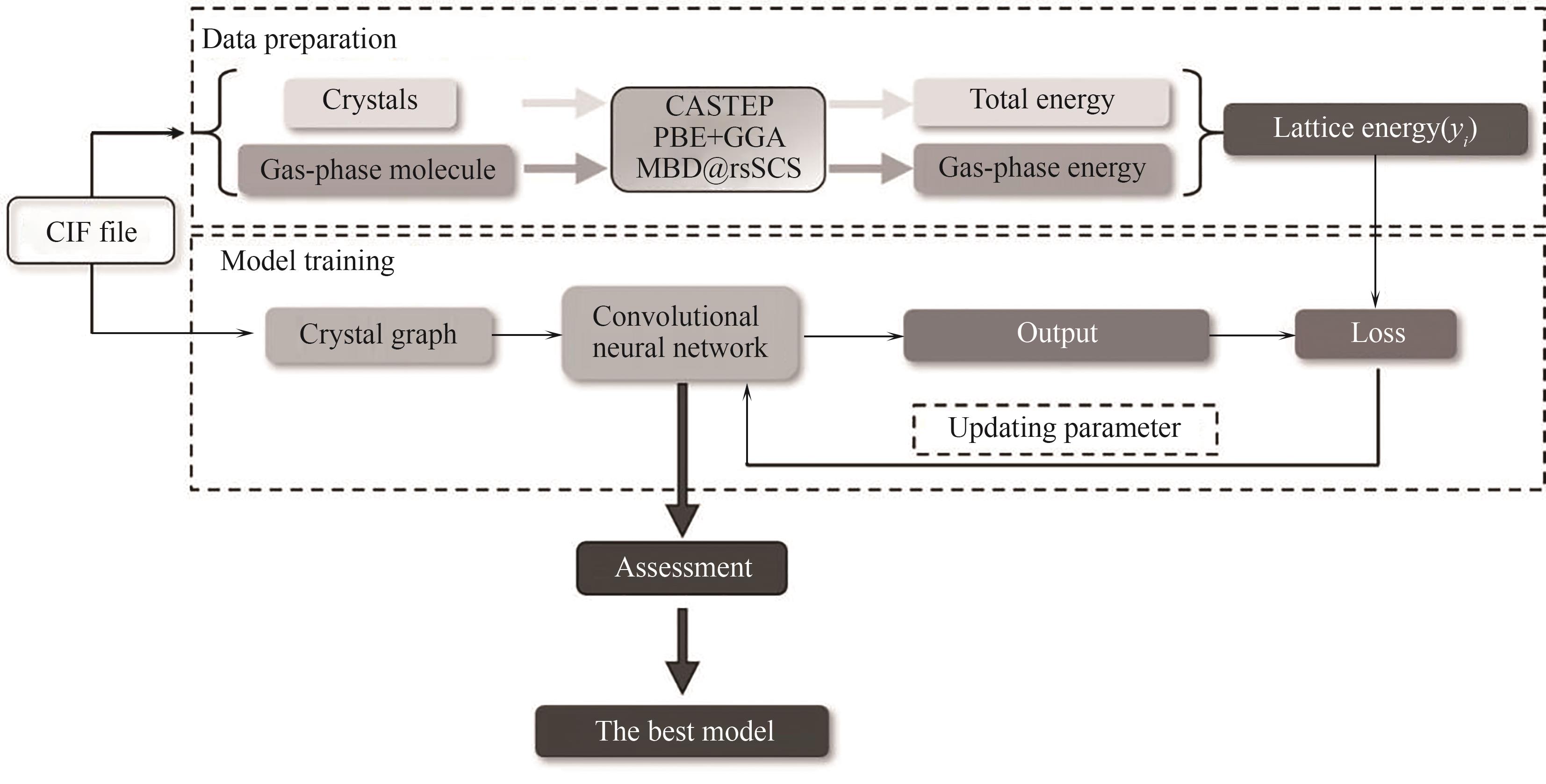
Fig.1 Basic framework of lattice energy regression model based on crystal graph convolutional neural network
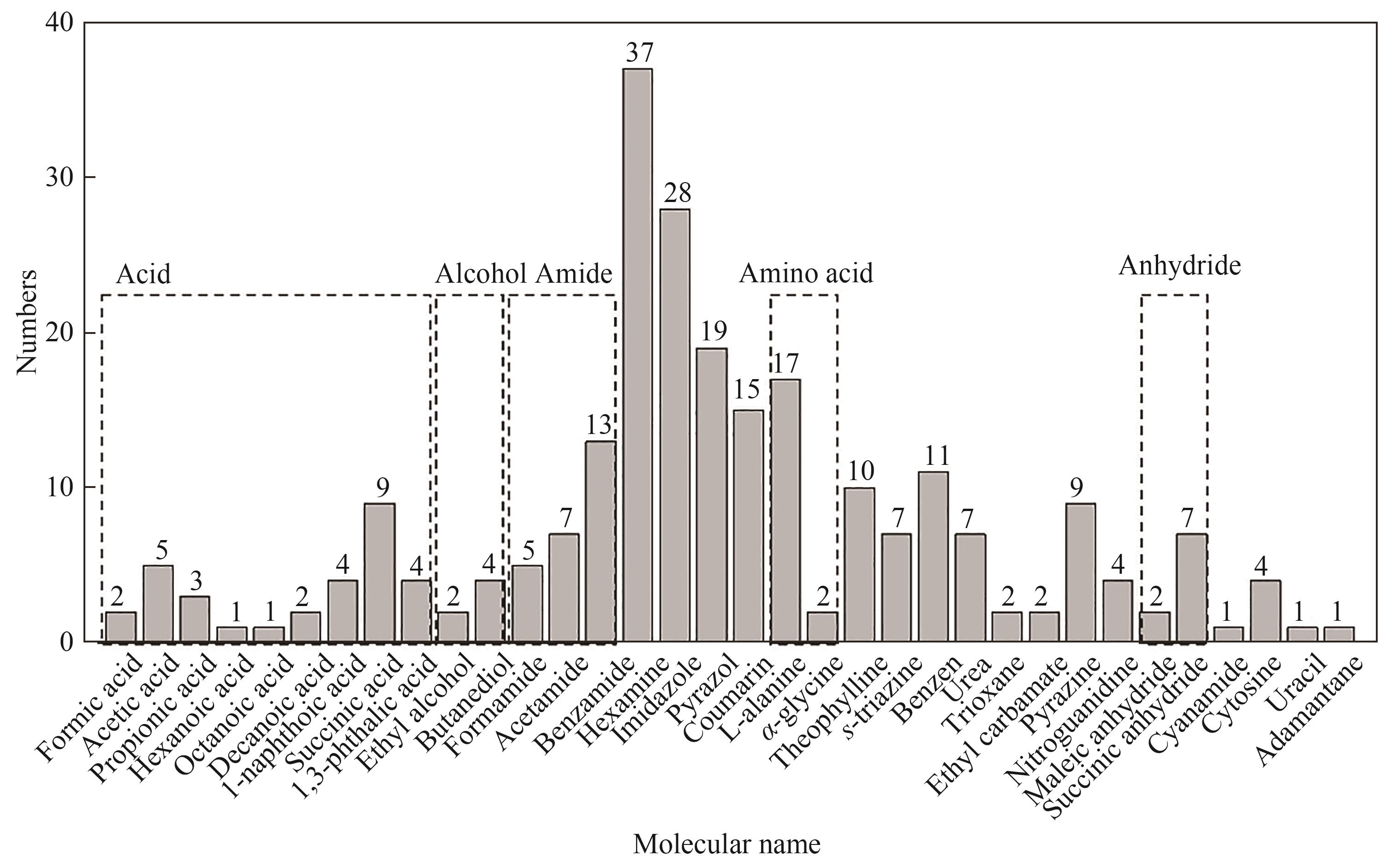
Fig.2 The distribution of data points: compounds with different functional groups, such as acids, alcohols, amides, amino acids and anhydrides, as well as different crystal structures of the same molecule
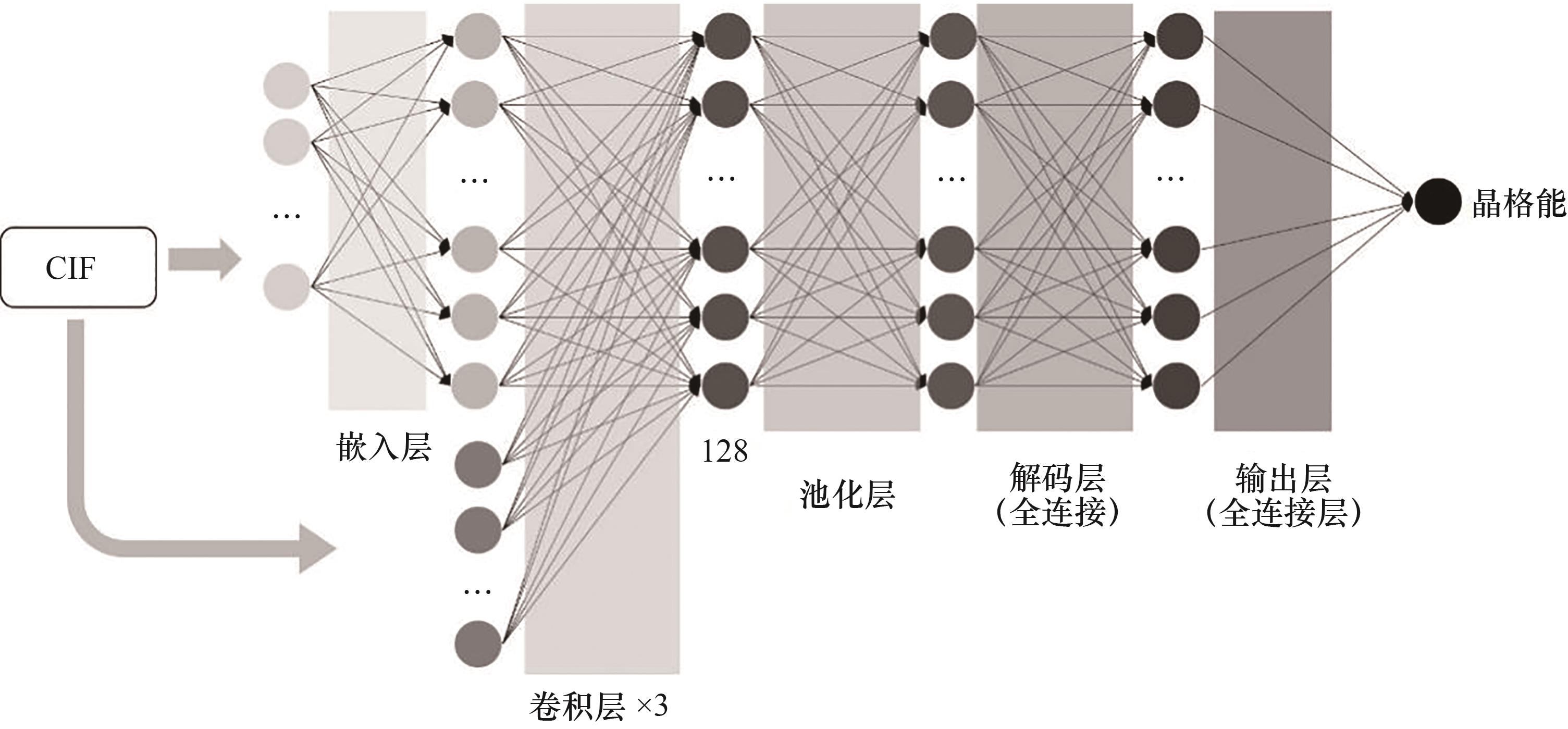
Fig.3 Structure diagram of CGCNN neural network
| 参数 | 设定值 |
|---|---|
| 批量大小 | 8 |
| 周期 | 800 |
| 学习率 | 0.01(动态) |
| 损失函数 | MSELoss |
| 优化程序 | SGD |
| 卷积层数 | 3 |
| 卷积层神经元数 | 128 |
| 激活函数 | SoftPlus |
Table 1 Model training parameter setting
| 参数 | 设定值 |
|---|---|
| 批量大小 | 8 |
| 周期 | 800 |
| 学习率 | 0.01(动态) |
| 损失函数 | MSELoss |
| 优化程序 | SGD |
| 卷积层数 | 3 |
| 卷积层神经元数 | 128 |
| 激活函数 | SoftPlus |
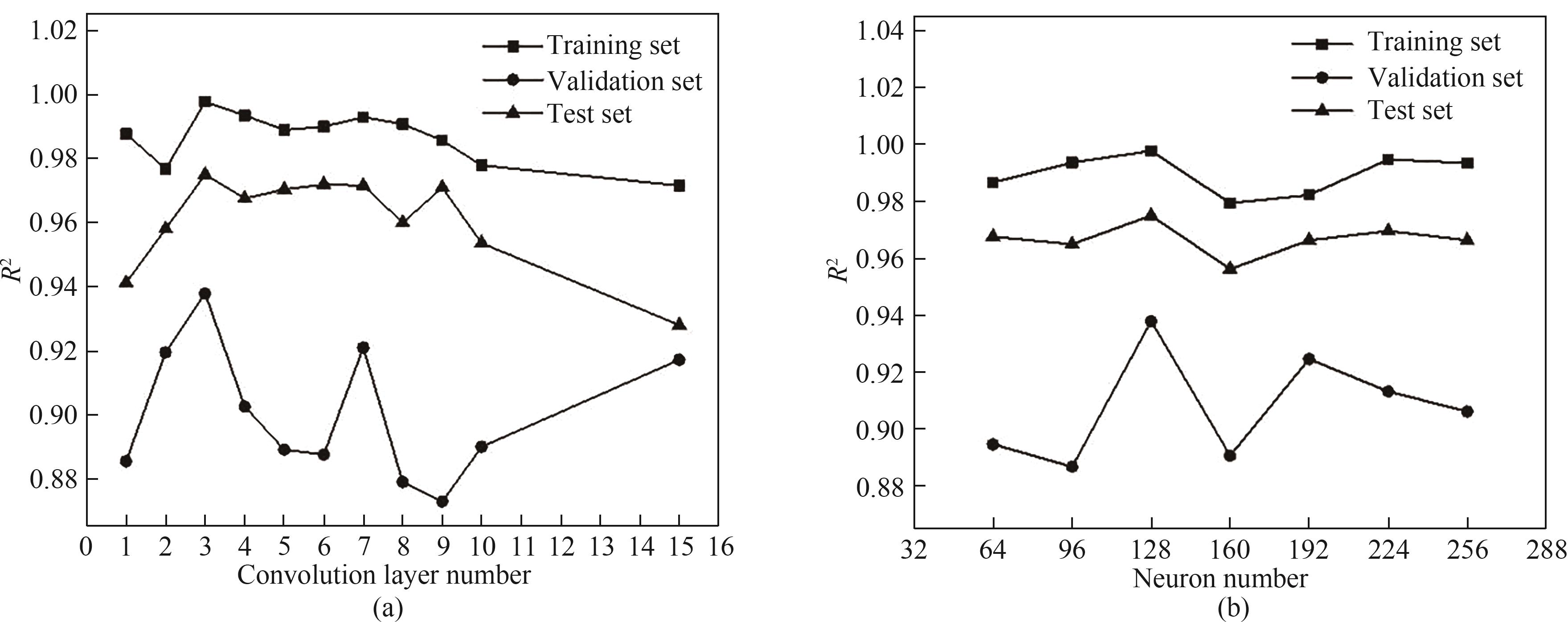
Fig.4 Performance of training set, validation set and test set under different hyperparameters: (a) performance curve of the model when the number of convolution layers is 1—15; (b) performance curve of the model when the number of neurons is 64—256
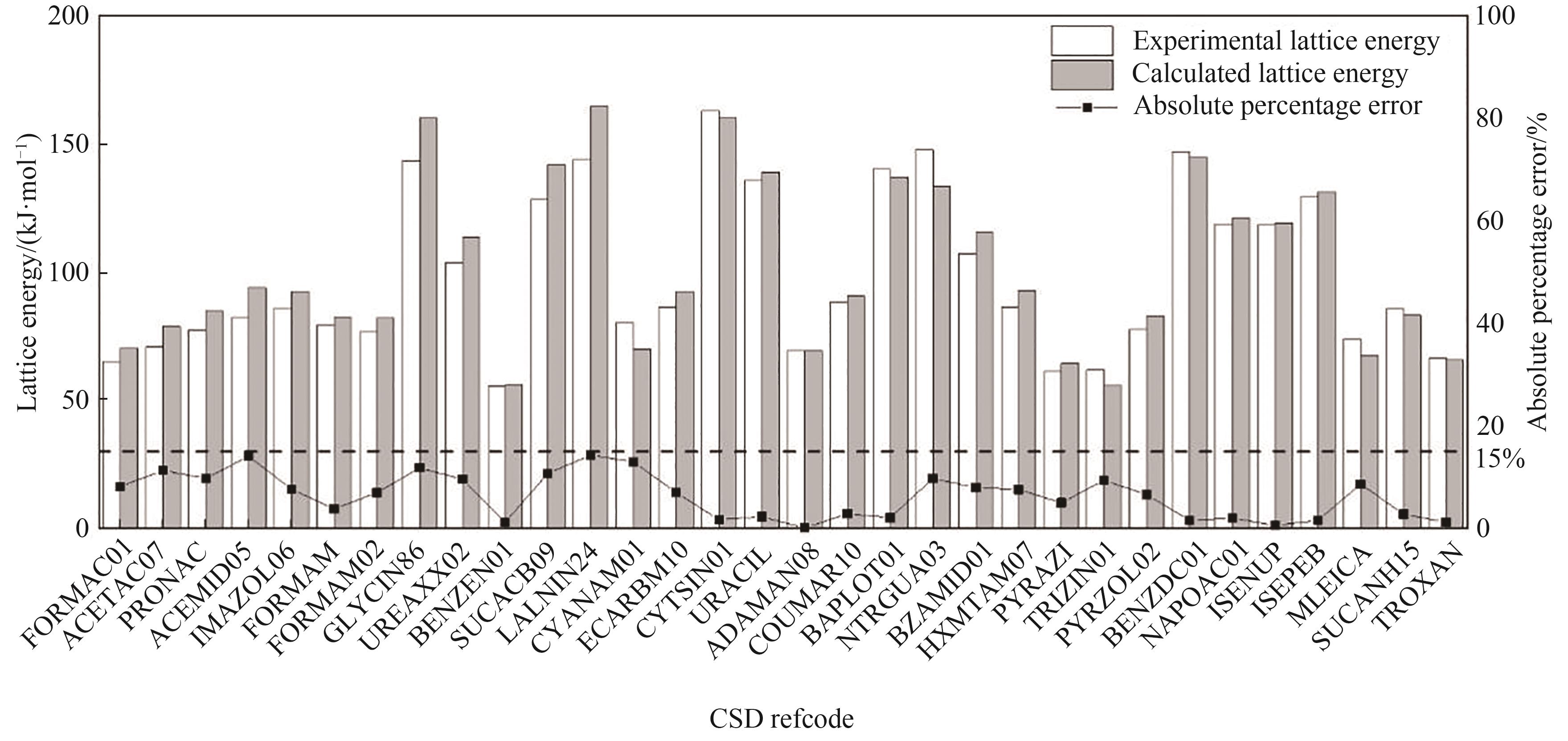
Fig.5 Comparison of experimental lattice energy with calculated lattice energy of 32 crystal structures
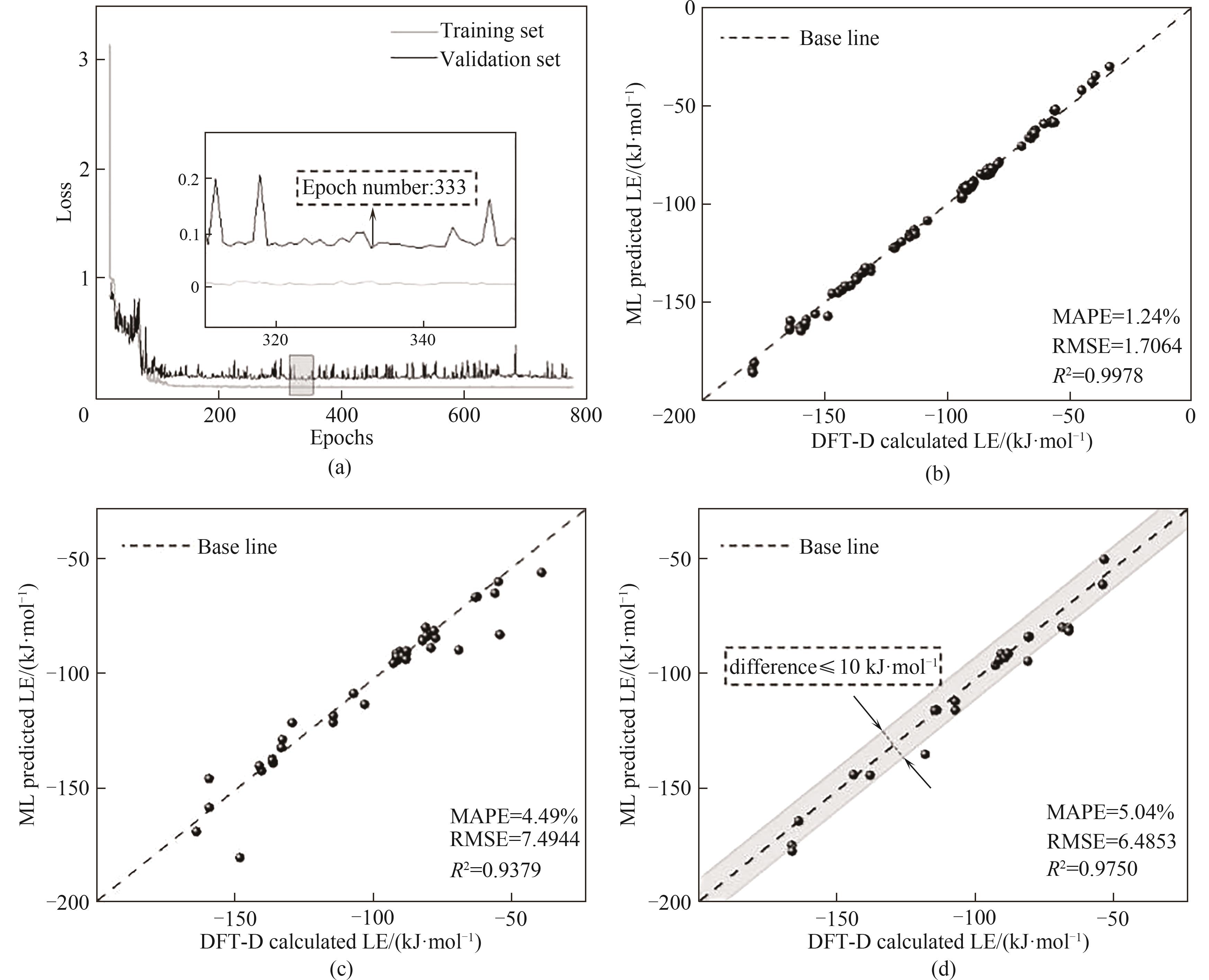
Fig.6 Model training results: (a) loss decline curve, the minimum loss is achieved at the 333rd epoch; (b) training set results; (c) validation set results; (d) test set results, the shaded area indicates the range where the difference between the calculated and predicted values of the lattice energy is less than or equal to 10 kJ·mol-1
| 1 | 贾凤. 全球价值链视角下中国医药产业国际竞争力研究[D]. 昆明: 昆明理工大学, 2022. |
| Jia F. Research on the international competitiveness of China pharmaceutical industry: from the perspective of global value chain[D]. Kunming: Kunming University of Science and Technology, 2022. | |
| 2 | 梁作中. 基于溶质-溶剂物系分子力场的可控结晶行为及关键调控机理研究[D]. 北京: 北京化工大学, 2016. |
| Liang Z Z. Study on the controllable crystallization behaviour and key regulation mechanism based on solute-solvent system and molecular force field[D]. Beijing: Beijing University of Chemical Technology, 2016. | |
| 3 | Lee E H. A practical guide to pharmaceutical polymorph screening & selection[J]. Asian Journal of Pharmaceutical Sciences, 2014, 9(4): 163-175. |
| 4 | Higashi K, Ueda K, Moribe K. Recent progress of structural study of polymorphic pharmaceutical drugs[J]. Advanced Drug Delivery Reviews, 2017, 117: 71-85. |
| 5 | Wang Y C, Lv J, Gao P Y, et al. Crystal structure prediction via efficient sampling of the potential energy surface[J]. Accounts of Chemical Research, 2022, 55(15): 2068-2076. |
| 6 | Guo M S, Sun X J, Chen J, et al. Pharmaceutical cocrystals: a review of preparations, physicochemical properties and applications[J]. Acta Pharmaceutica Sinica B, 2021, 11(8): 2537-2564. |
| 7 | Singhal D, Curatolo W. Drug polymorphism and dosage form design: a practical perspective[J]. Advanced Drug Delivery Reviews, 2004, 56(3): 335-347. |
| 8 | Singh M K. Predicting lattice energy and structure of molecular crystals by first-principles method: role of dispersive interactions[J]. Journal of Crystal Growth, 2014, 396: 14-23. |
| 9 | Ropers J, Mosca M M, Anosova O, et al. Fast predictions of lattice energies by continuous isometry invariants of crystal structures[C]// Data Analytics and Management in Data Intensive Domains. Cham: Springer International Publishing, 2022: 178-192. |
| 10 | Nyman J, Day G M. Static and lattice vibrational energy differences between polymorphs[J]. CrystEngComm, 2015, 17(28): 5154-5165. |
| 11 | Geatches D, Rosbottom I, Marchese Robinson R L, et al. Off-the-shelf DFT-DISPersion methods: are they now “on-trend” for organic molecular crystals?[J]. The Journal of Chemical Physics, 2019, 151(4): 044106. |
| 12 | Feng S X, Li T L. Predicting lattice energy of organic crystals by density functional theory with empirically corrected dispersion energy[J]. Journal of Chemical Theory and Computation, 2006, 2(1): 149-156. |
| 13 | Fang T, Li W, Gu F W, et al. Accurate prediction of lattice energies and structures of molecular crystals with molecular quantum chemistry methods[J]. Journal of Chemical Theory and Computation, 2015, 11(1): 91-98. |
| 14 | Zheng Z Y, Zhao J J, Sun Y Y, et al. Structures and lattice energies of molecular crystals using density functional theory: assessment of a local atomic potential approach[J]. Chemical Physics Letters, 2012, 550: 94-98. |
| 15 | Mortazavi M, Hoja J, Aerts L, et al. Computational polymorph screening reveals late-appearing and poorly-soluble form of rotigotine[J]. Communications Chemistry, 2019, 2: 70. |
| 16 | Pan J. Scaling up system size in materials simulation[J]. Nature Computational Science, 2021, 1(2): 95. |
| 17 | Verma P, Truhlar D G. Status and challenges of density functional theory[J]. Trends in Chemistry, 2020, 2(4): 302-318. |
| 18 | Pugliese R, Regondi S, Marini R. Machine learning-based approach: global trends, research directions, and regulatory standpoints[J]. Data Science and Management, 2021, 4: 19-29. |
| 19 | Sarker I H. Machine learning: algorithms, real-world applications and research directions[J]. Computer Science, 2021, 2(3): 160. |
| 20 | Xu P C, Chen H M, Li M J, et al. New opportunity: machine learning for polymer materials design and discovery[J]. Advanced Theory and Simulations, 2022, 5(5): 2100565. |
| 21 | Huang G N, Guo Y N, Chen Y, et al. Application of machine learning in material synthesis and property prediction[J]. Materials, 2023, 16(17): 5977. |
| 22 | Xie T, Grossman J C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties[J]. Physical Review Letters, 2018, 120(14): 145301. |
| 23 | Cheng G J, Gong X G, Yin W J. Crystal structure prediction by combining graph network and optimization algorithm[J]. Nature Communications, 2022, 13(1): 1492. |
| 24 | Kusaba M, Liu C, Yoshida R. Crystal structure prediction with machine learning-based element substitution[J]. Computational Materials Science, 2022, 211: 111496. |
| 25 | Buchholz H K, Stein M. Accurate lattice energies of organic molecular crystals from periodic turbomole calculations[J]. Journal of Computational Chemistry, 2018, 39(19): 1335-1343. |
| 26 | 赵慧茹. 量子化学方法在分子晶体计算中的应用[D]. 南京: 南京大学, 2015. |
| Zhao H R. The application of quantum chemical methods in calculation of molecular crystals[D]. Nanjing: Nanjing University, 2015. | |
| 27 | 王恋. 分子间氢键和色散力对含氧芳香族化合物激发态动力学的影响[D]. 武汉: 中国科学院大学(中国科学院武汉物理与数学研究所), 2020. |
| Wang L. Effect of intermolecular hydrogen bonding and dispersion interactions on excited state dynamics of oxygen-containing aromatic compounds[D]. Wuhan: University of Chinese Academy of Sciences(Wuhan Institute of Physics and Mathematics of Chinese Academy of Sciences), 2020. | |
| 28 | Dolgonos G A, Boese A D. Adjusting dispersion parameters for the density-functional tight-binding description of molecular crystals[J]. Chemical Physics Letters, 2019, 718: 7-11. |
| 29 | Wei J, Chu X, Sun X Y, et al. Machine learning in materials science[J]. InfoMat, 2019, 1(3): 338-358. |
| 30 | Kaundinya P R, Choudhary K, Kalidindi S R. Machine learning approaches for feature engineering of the crystal structure: application to the prediction of the formation energy of cubic compounds[J]. Physical Review Materials, 2021, 5(6): 063802. |
| 31 | Feng S, Zhou H Y, Dong H B. Application of deep transfer learning to predicting crystal structures of inorganic substances[J]. Computational Materials Science, 2021, 195: 110476. |
| 32 | Ong S P, Richards W D, Jain A, et al. Python materials genomics (pymatgen): a robust, open-source python library for materials analysis[J]. Computational Materials Science, 2013, 68: 314-319. |
| 33 | Başar M S, Küçükönder H. Measuring the correlation between commercial and economic states of countries (B2G relations) and the E-government readiness index by using neural networks[J]. Open Journal of Business and Management, 2014, 2(2): 110-115. |
| [1] | Shaoji WANG, Kuangrong HAO, Lei CHEN. Research on temperature forecasting of polyester fiber esterification process based on federated learning [J]. CIESC Journal, 2025, 76(1): 283-295. |
| [2] | Han ZHANG, Shuning ZHANG, Ke LIU, Guanlong DENG. Particle size prediction of cobalt oxalate synthesis process based on slow feature analysis and least squares support vector regression [J]. CIESC Journal, 2024, 75(6): 2313-2321. |
| [3] | Sirui CHEN, Jingliang BI, Lei WANG, Yuanyuan LI, Gui LU. Unsupervised-feature extraction of gas-liquid two-phase flow pattern based on convolutional autoencoder: principle and application [J]. CIESC Journal, 2024, 75(3): 847-857. |
| [4] | Yongjun XIAO, Zhaochong SHI, Ren WAN, Fan SONG, Changjun PENG, Honglai LIU. Prediction of self-diffusion coefficients of ionic liquids using back-propagation neural networks [J]. CIESC Journal, 2024, 75(2): 429-438. |
| [5] | Linqi YAN, Zhenlei WANG. Multi-step predictive soft sensor modeling based on STA-BiLSTM-LightGBM combined model [J]. CIESC Journal, 2023, 74(8): 3407-3418. |
| [6] | Gang YIN, Yihui LI, Fei HE, Wenqi CAO, Min WANG, Feiya YAN, Yu XIANG, Jian LU, Bin LUO, Runting LU. Early warning method of aluminum reduction cell leakage accident based on KPCA and SVM [J]. CIESC Journal, 2023, 74(8): 3419-3428. |
| [7] | Xuejin GAO, Yuzhuo YAO, Huayun HAN, Yongsheng QI. Fault monitoring of fermentation process based on attention dynamic convolutional autoencoder [J]. CIESC Journal, 2023, 74(6): 2503-2521. |
| [8] | Cheng YUN, Qianlin WANG, Feng CHEN, Xin ZHANG, Zhan DOU, Tingjun YAN. Deep-mining risk evolution path of chemical processes based on community structure [J]. CIESC Journal, 2023, 74(4): 1639-1650. |
| [9] | Xinyuan WU, Qilei LIU, Boyuan CAO, Lei ZHANG, Jian DU. Group2vec: group vector representation and its property prediction applications based on unsupervised machine learning [J]. CIESC Journal, 2023, 74(3): 1187-1194. |
| [10] | Xiangyu LI, Lin SUI, Junxia MA, Weili XIONG. ONLSTM soft sensor modeling based on time series transfer and dual stream weighting [J]. CIESC Journal, 2023, 74(11): 4622-4633. |
| [11] | Xuejin GAO, Kun CHENG, Huayun HAN, Huihui Gao, Yongsheng QI. Fault diagnosis of chillers using central loss conditional generative adversarial network [J]. CIESC Journal, 2022, 73(9): 3950-3962. |
| [12] | Le ZHOU, Chengkai SHEN, Chao WU, Beiping HOU, Zhihuan SONG. Deep fusion feature extraction network and its application in chemical process soft sensing [J]. CIESC Journal, 2022, 73(7): 3156-3165. |
| [13] | Zihao QI, Wenqi ZHONG, Xi CHEN, Guanwen ZHOU, Xiaoliang ZHAO, Meijing XIN, Yi CHEN, Yongchang ZHU. Research on dynamic characteristics of cement raw meal decomposition process based on hybrid modeling [J]. CIESC Journal, 2022, 73(5): 2039-2051. |
| [14] | Jiahui REN, Yu LIU, Chao LIU, Lang LIU, Ying LI. Critical temperature prediction of working fluids using molecular fingerprints and topological indices [J]. CIESC Journal, 2022, 73(4): 1493-1500. |
| [15] | Zhibin LU, Yimeng LI, Chang HE, Bingjian ZHANG, Qinglin CHEN, Ming PAN. Integrating physics-informed neural networks with partitioned coupling strategy for modeling conjugate heat transfer [J]. CIESC Journal, 2022, 73(12): 5483-5493. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

