CIESC Journal ›› 2024, Vol. 75 ›› Issue (4): 1241-1255.DOI: 10.11949/0438-1157.20231030
• Reviews and monographs • Previous Articles Next Articles
Dongfei LIU( ), Fan ZHANG, Zheng LIU, Diannan LU()
), Fan ZHANG, Zheng LIU, Diannan LU()
Received:2023-10-07
Revised:2024-02-20
Online:2024-06-06
Published:2024-04-25
Contact:
Diannan LU
刘东飞(), 张帆, 刘铮, 卢滇楠()
通讯作者:
卢滇楠
作者简介:刘东飞 (1999—),男,博士研究生,ldf20@mails.tsinghua.edu.cn
基金资助:CLC Number:
Dongfei LIU, Fan ZHANG, Zheng LIU, Diannan LU. A review of machine learning potentials and their applications to molecular simulation[J]. CIESC Journal, 2024, 75(4): 1241-1255.
刘东飞, 张帆, 刘铮, 卢滇楠. 机器学习势及其在分子模拟中的应用综述[J]. 化工学报, 2024, 75(4): 1241-1255.
Add to citation manager EndNote|Ris|BibTeX
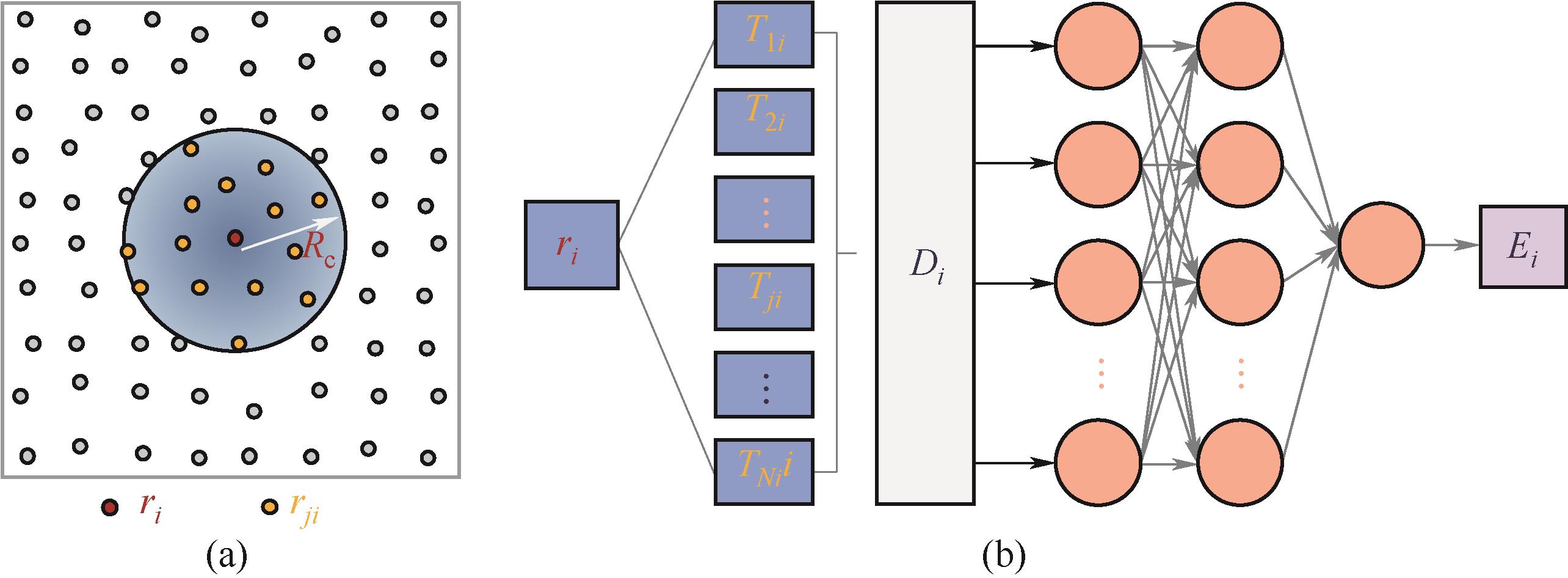
Fig.1 (a) Locality approximation for central atom and neighboring atoms within the cutoff radius Rc; (b) Under the locality approximation, each atom corresponds to one separate neural network
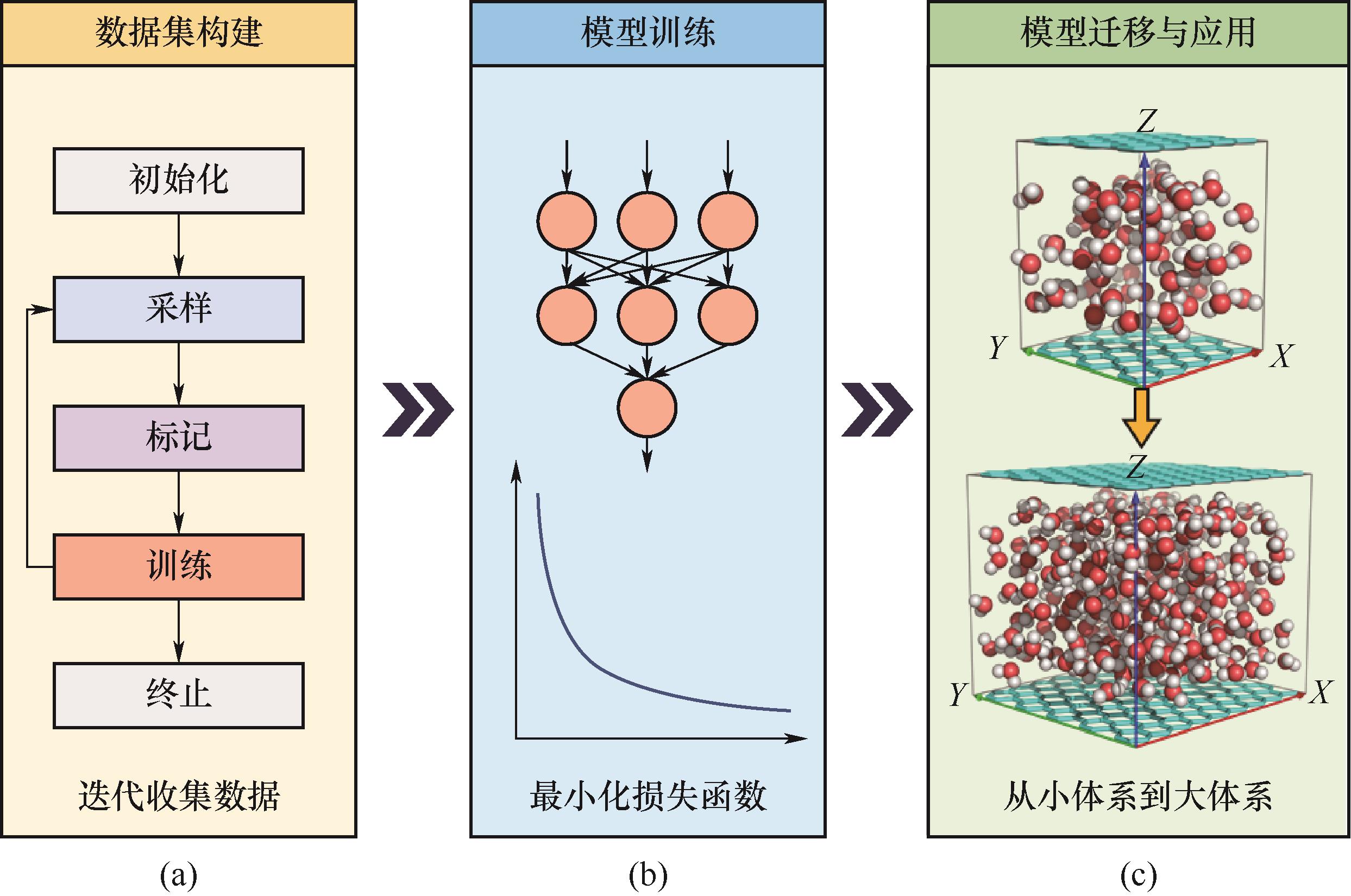
Fig.2 The general process of training and applying MLPs
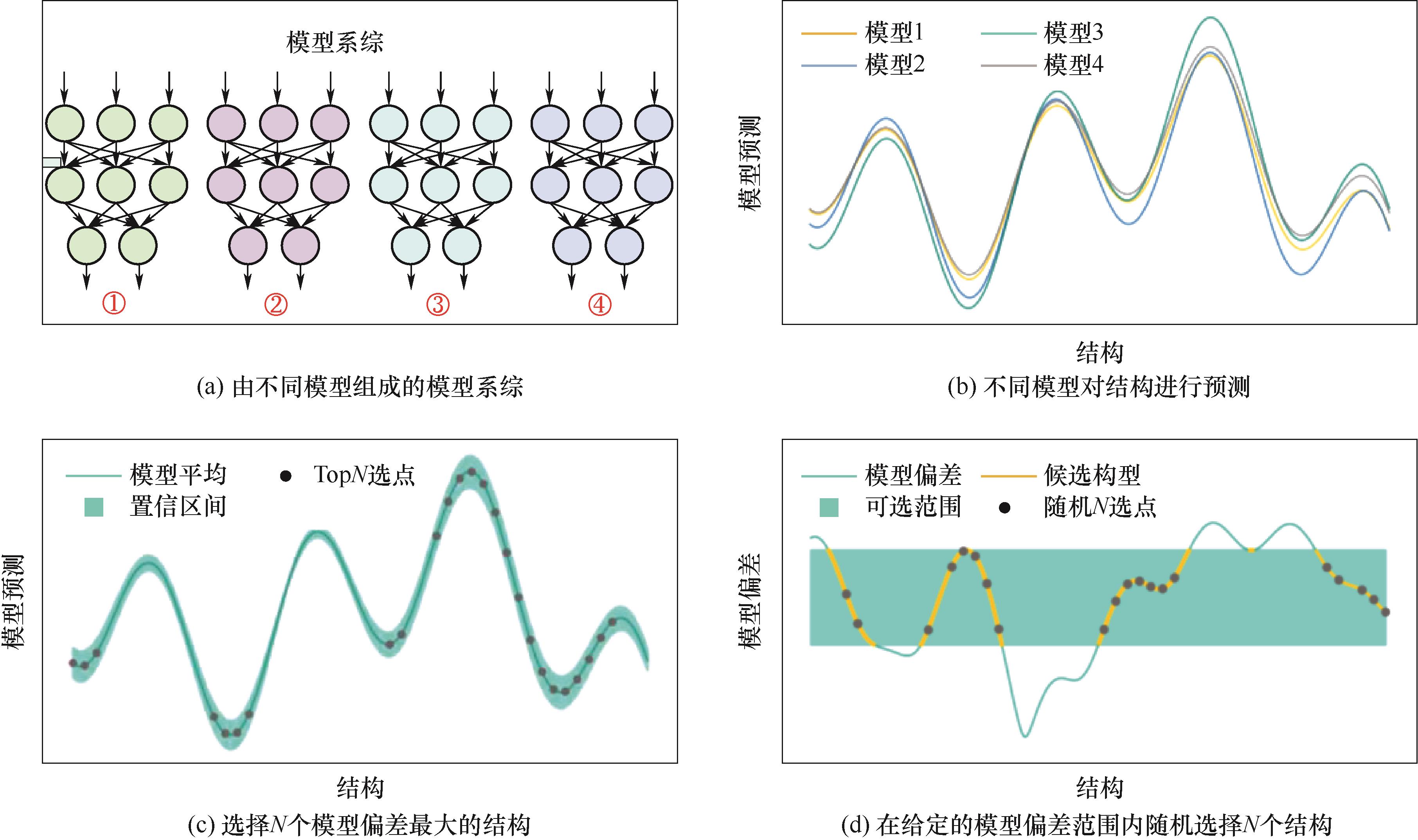
Fig.3 The sampling process in active learning
| 机器学习方法 | MLPs模型 | 模型特点 | 应用体系 |
|---|---|---|---|
| 神经网络 | DPMD[ | 二代NNPs | 水的相图预测[ |
| ANI[ | 二代NNPs | ANI-1x数据库[ | |
| TensorMol[ | 三代NNPs | 水分子簇[ | |
| n2p2[ | 二代NNPs | 体相水[ | |
| RuNNer[ | 四代NNPs | 水在ZnO表面的解离[ | |
| EANN[ | 二代NNPs | 有机小分子与Cu、Ge等周期性体系[ | |
| DTNN[ | 消息传递机制 | GDB-7、GDB-9[ | |
| SchNet[ | 消息传递机制 | QM9[ | |
| DimeNet[ | 消息传递机制 | QM9[ | |
| GemNet[ | 消息传递机制 | COLL[ | |
| NequIP[ | 消息传递机制 | MD17[ | |
| Allegro[ | 消息传递机制 | QM9[ | |
| MACE[ | 消息传递机制 | MD17[ | |
| ViSNet[ | 消息传递机制 | QM9[ | |
| HIP-NN[ | 二代NNPs | QM9[ | |
| SpookyNet[ | 四代NNPs | C10H2/ C10H | |
| PhysNet[ | 三代NNPs | QM9[ | |
| ForceNet[ | 消息传递机制 | OC20[ | |
| 核岭回归 | GDML[ | 核方法 | MD17[ |
| sGDML[ | 核方法 | 有机小分子[ | |
| 高斯过程回归 | GAP[ | 核方法 | 单质体系[ |
Table 1 Summary of MLPs models, model characteristics, and typical cases of each model
| 机器学习方法 | MLPs模型 | 模型特点 | 应用体系 |
|---|---|---|---|
| 神经网络 | DPMD[ | 二代NNPs | 水的相图预测[ |
| ANI[ | 二代NNPs | ANI-1x数据库[ | |
| TensorMol[ | 三代NNPs | 水分子簇[ | |
| n2p2[ | 二代NNPs | 体相水[ | |
| RuNNer[ | 四代NNPs | 水在ZnO表面的解离[ | |
| EANN[ | 二代NNPs | 有机小分子与Cu、Ge等周期性体系[ | |
| DTNN[ | 消息传递机制 | GDB-7、GDB-9[ | |
| SchNet[ | 消息传递机制 | QM9[ | |
| DimeNet[ | 消息传递机制 | QM9[ | |
| GemNet[ | 消息传递机制 | COLL[ | |
| NequIP[ | 消息传递机制 | MD17[ | |
| Allegro[ | 消息传递机制 | QM9[ | |
| MACE[ | 消息传递机制 | MD17[ | |
| ViSNet[ | 消息传递机制 | QM9[ | |
| HIP-NN[ | 二代NNPs | QM9[ | |
| SpookyNet[ | 四代NNPs | C10H2/ C10H | |
| PhysNet[ | 三代NNPs | QM9[ | |
| ForceNet[ | 消息传递机制 | OC20[ | |
| 核岭回归 | GDML[ | 核方法 | MD17[ |
| sGDML[ | 核方法 | 有机小分子[ | |
| 高斯过程回归 | GAP[ | 核方法 | 单质体系[ |
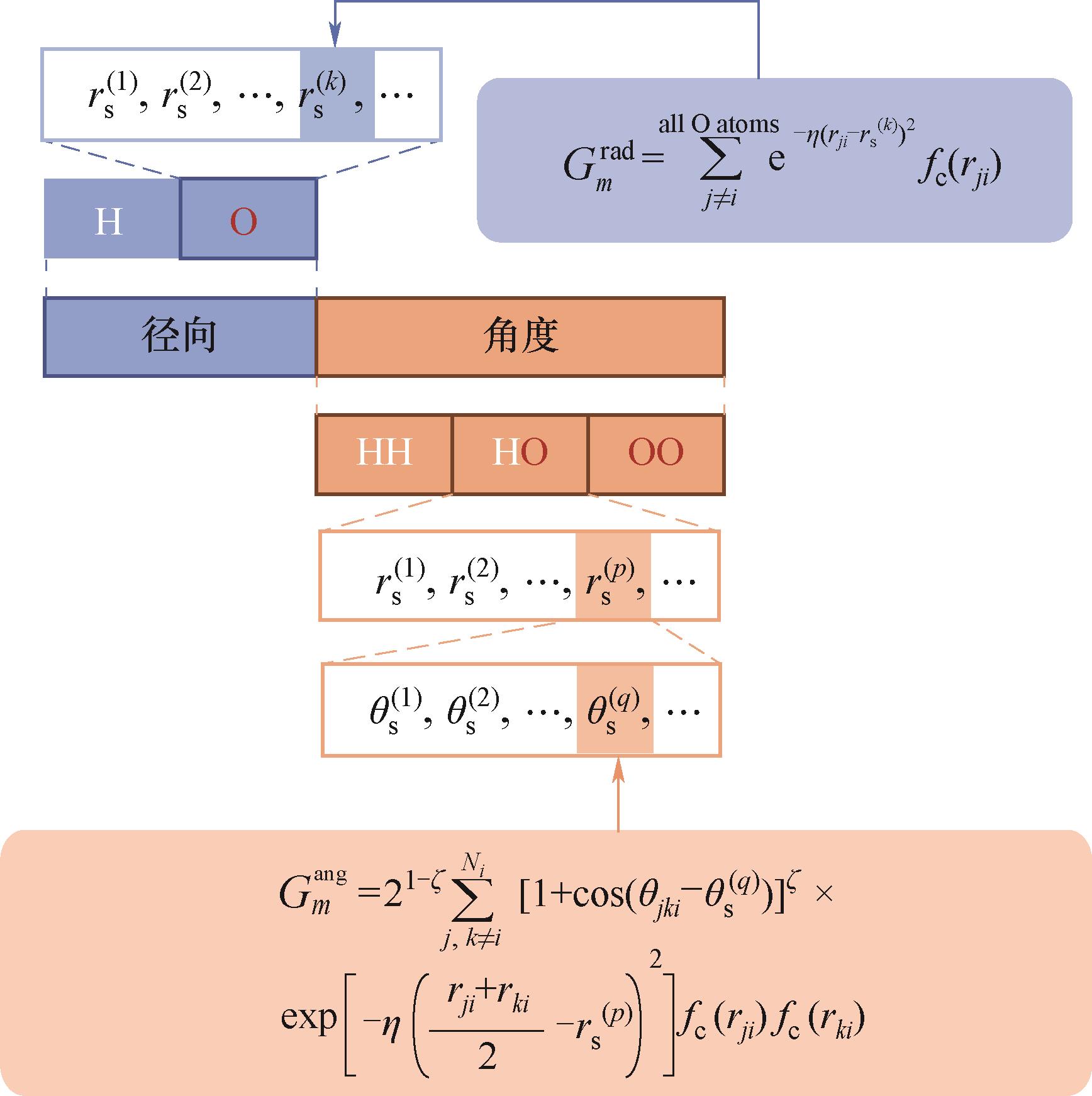
Fig.4 The construction of ACSF descriptors for water molecules system in ANI model
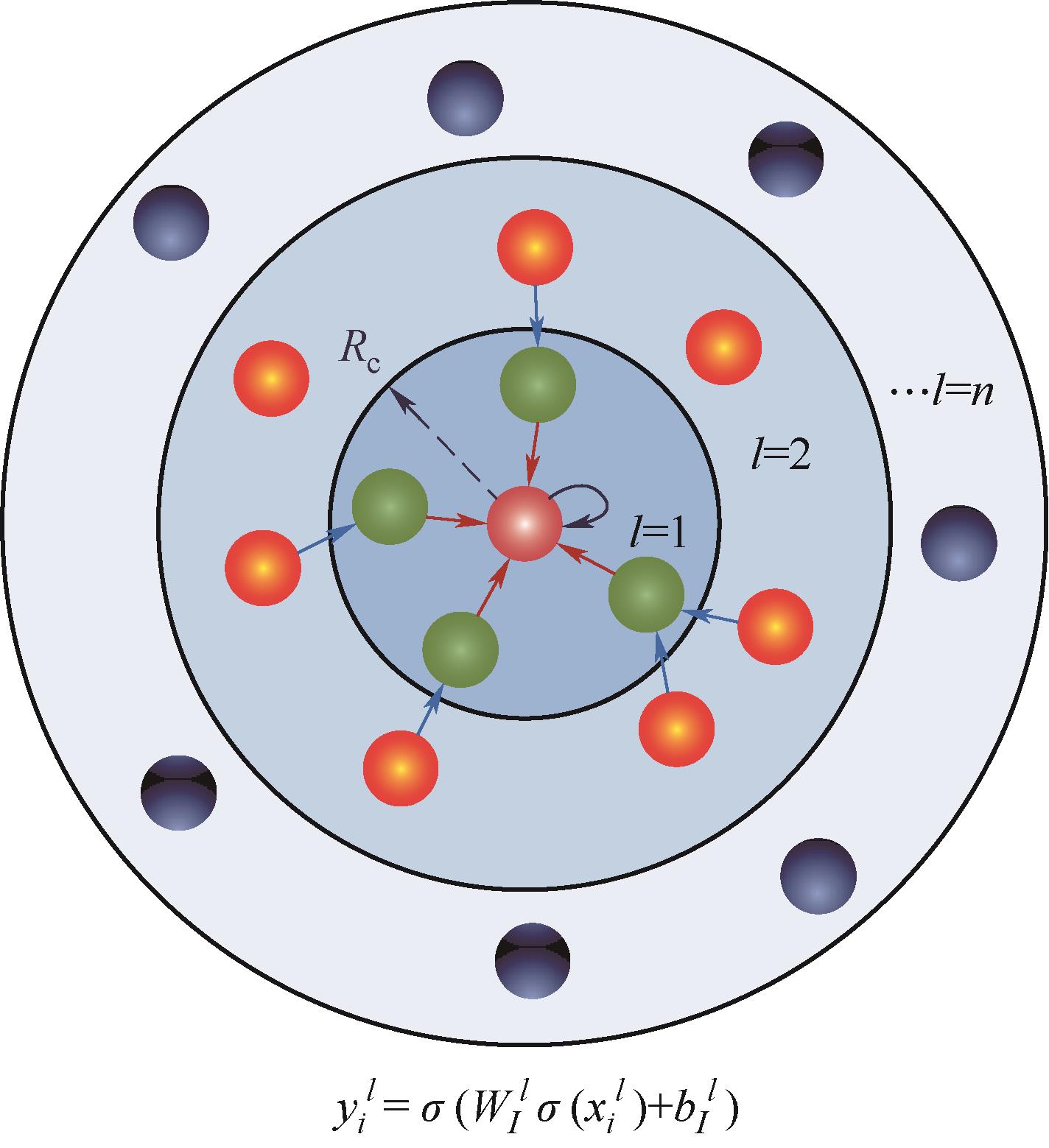
Fig.5 Scheme of the message passing process in graph neural network
| 1 | Rahman A, Stillinger F H. Molecular dynamics study of liquid water[J]. The Journal of Chemical Physics, 1971, 55(7): 3336-3359. |
| 2 | Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory[J]. Physical Review Letters, 1985, 55(22): 2471-2474. |
| 3 | Schulz R, Lindner B, Petridis L, et al. Scaling of multimillion-atom biological molecular dynamics simulation on a petascale supercomputer[J]. Journal of Chemical Theory and Computation, 2009, 5(10): 2798-2808. |
| 4 | Fu Y, Bernasconi L, Liu P. Ab initio molecular dynamics simulations of the SN1/SN2 mechanistic continuum in glycosylation reactions[J]. Journal of the American Chemical Society, 2021, 143(3): 1577-1589. |
| 5 | Sangiovanni D G, Gueorguiev G K, Kakanakova-Georgieva A. Ab initio molecular dynamics of atomic-scale surface reactions: insights into metal organic chemical vapor deposition of AlN on graphene[J]. Physical Chemistry Chemical Physics: PCCP, 2018, 20(26): 17751-17761. |
| 6 | Kocer E, Ko T W, Behler J. Neural network potentials: a concise overview of methods[J]. Annual Review of Physical Chemistry, 2022, 73: 163-186. |
| 7 | Cramer C J. Essentials of Computational Chemistry: Theories and Models[M]. 2nd ed. Chichester, West Sussex, England: Wiley, 2004:6. |
| 8 | Weiner S J, Kollman P A, Case D A, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins[J]. Journal of the American Chemical Society, 1984, 106(3): 765-784. |
| 9 | Weinan E. The dawning of a new era in applied mathematics[J]. Notices of the American Mathematical Society, 2021, 68(4): 1. |
| 10 | Hornik K, Stinchcombe M, White H. Multilayer feedforward networks are universal approximators[J]. Neural Networks, 1989, 2(5): 359-366. |
| 11 | Blank T B, Brown S D, Calhoun A W, et al. Neural network models of potential energy surfaces[J]. The Journal of Chemical Physics, 1995, 103(10): 4129-4137. |
| 12 | Behler J, Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces[J]. Physical Review Letters, 2007, 98(14): 146401. |
| 13 | Wang H, Zhang L F, Han J Q, et al. DeePMD-kit: a deep learning package for many-body potential energy representation and molecular dynamics[J]. Computer Physics Communications, 2018, 228: 178-184. |
| 14 | Schütt K T, Kessel P, Gastegger M, et al. SchNetPack: a deep learning toolbox for atomistic systems[J]. Journal of Chemical Theory and Computation, 2019, 15(1): 448-455. |
| 15 | Schütt K T, Hessmann S S P, Gebauer N W A, et al. SchNetPack 2.0: a neural network toolbox for atomistic machine learning[EB/OL]. 2022. . |
| 16 | Batzner S, Musaelian A, Sun L X, et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials[J]. Nature Communications, 2022, 13: 2453. |
| 17 | Hu W H, Shuaibi M, Das A, et al. ForceNet: a graph neural network for large-scale quantum calculations[EB/OL]. 2021. . |
| 18 | Gasteiger J, Yeshwanth C, Günnemann S. Directional message passing on molecular graphs via synthetic coordinates[EB/OL]. 2021. . |
| 19 | Gasteiger J, Becker F, Günnemann S. GemNet: universal directional graph neural networks for molecules[EB/OL]. 2021. . |
| 20 | Chmiela S, Sauceda H E, Poltavsky I, et al. sGDML: constructing accurate and data efficient molecular force fields using machine learning[J]. Computer Physics Communications, 2019, 240: 38-45. |
| 21 | Chmiela S, Tkatchenko A, Sauceda H E, et al. Machine learning of accurate energy-conserving molecular force fields[J]. Science Advances, 2017, 3(5): e1603015. |
| 22 | Chmiela S, Sauceda H E, Müller K R, et al. Towards exact molecular dynamics simulations with machine-learned force fields[J]. Nature Communications, 2018, 9: 3887. |
| 23 | Chmiela S, Vassilev-Galindo V, Unke O T, et al. Accurate global machine learning force fields for molecules with hundreds of atoms[J]. Science Advances, 2023, 9(2): eadf0873. |
| 24 | Bartók A P, Payne M C, Kondor R, et al. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons[J]. Physical Review Letters, 2010, 104(13): 136403. |
| 25 | Bartók A P, Csányi G. Gaussian approximation potentials: a brief tutorial introduction[J]. International Journal of Quantum Chemistry, 2015, 115(16): 1051-1057. |
| 26 | Thompson A P, Swiler L P, Trott C R, et al. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials[J]. Journal of Computational Physics, 2015, 285: 316-330. |
| 27 | Li X G, Hu C Z, Chen C, et al. Quantum-accurate spectral neighbor analysis potential models for Ni-Mo binary alloys and FCC metals[J]. Physical Review B, 2018, 98(9): 094104. |
| 28 | Rosenbrock C W, Gubaev K, Shapeev A V, et al. Machine-learned interatomic potentials for alloys and alloy phase diagrams[J]. NPJ Computational Materials, 2021, 7: 24. |
| 29 | Behler J. Four generations of high-dimensional neural network potentials[J]. Chemical Reviews, 2021, 121(16): 10037-10072. |
| 30 | Deringer V L, Csányi G. Machine learning based interatomic potential for amorphous carbon[J]. Physical Review B, 2017, 95(9): 094203. |
| 31 | Schran C, Thiemann F L, Rowe P, et al. Machine learning potentials for complex aqueous systems made simple[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(38): e2110077118. |
| 32 | Thiemann F L, Schran C, Rowe P, et al. Water flow in single-wall nanotubes: oxygen makes it slip, hydrogen makes it stick[J]. ACS Nano, 2022, 16(7): 10775-10782. |
| 33 | Yang M Y, Bonati L, Polino D, et al. Using metadynamics to build neural network potentials for reactive events: the case of urea decomposition in water[J]. Catalysis Today, 2022, 387: 143-149. |
| 34 | Zhao W, Qiu H, Guo W L. A deep neural network potential for water confined in graphene nanocapillaries[J]. The Journal of Physical Chemistry C, 2022, 126(25): 10546-10553. |
| 35 | Himanen L, Jäger M O J, Morooka E V, et al. DScribe: library of descriptors for machine learning in materials science[J]. Computer Physics Communications, 2020, 247: 106949. |
| 36 | Bartók A P, Kondor R, Csányi G. On representing chemical environments[J]. Physical Review B, 2013, 87(18): 184115. |
| 37 | Rupp M, Tkatchenko A, Müller K R, et al. Fast and accurate modeling of molecular atomization energies with machine learning[J]. Physical Review Letters, 2012, 108(5): 058301. |
| 38 | Jindal S, Chiriki S, Bulusu S S. Spherical harmonics based descriptor for neural network potentials: structure and dynamics of Au147 nanocluster[J]. The Journal of Chemical Physics, 2017, 146(20): 204301. |
| 39 | Rupp M. Unified representation of molecules and crystals for machine learning[J]. Machine Learning: Science and Technology, 2022, 3(4): 045017. |
| 40 | von Lilienfeld O A, Müller K R, Tkatchenko A. Exploring chemical compound space with quantum-based machine learning[J]. Nature Reviews Chemistry, 2020, 4: 347-358. |
| 41 | Schran C, Brezina K, Marsalek O. Committee neural network potentials control generalization errors and enable active learning[J]. The Journal of Chemical Physics, 2020, 153(10): 104105. |
| 42 | Zhang L F, Lin D Y, Wang H, et al. Active learning of uniformly accurate interatomic potentials for materials simulation[J]. Physical Review Materials, 2019, 3(2): 023804. |
| 43 | Podryabinkin E V, Tikhonov E V, Shapeev A V, et al. Accelerating crystal structure prediction by machine-learning interatomic potentials with active learning[J]. Physical Review B, 2019, 99(6): 064114. |
| 44 | Bernstein N, Csányi G, Deringer V L. De novo exploration and self-guided learning of potential-energy surfaces[J]. NPJ Computational Materials, 2019, 5: 99. |
| 45 | Schran C, Behler J, Marx D. Automated fitting of neural network potentials at coupled cluster accuracy: protonated water clusters as testing ground[J]. Journal of Chemical Theory and Computation, 2020, 16(1): 88-99. |
| 46 | Tan A R, Urata S, Goldman S, et al. Single-model uncertainty quantification in neural network potentials does not consistently outperform model ensembles[J]. NPJ Computational Materials, 2023, 9: 225. |
| 47 | Zhang Y Z, Wang H D, Chen W J, et al. DP-GEN: a concurrent learning platform for the generation of reliable deep learning based potential energy models[J]. Computer Physics Communications, 2020, 253: 107206. |
| 48 | Smith J S, Nebgen B, Lubbers N, et al. Less is more: sampling chemical space with active learning[J]. The Journal of Chemical Physics, 2018, 148(24): 241733. |
| 49 | Unke O T, Meuwly M. PhysNet: a neural network for predicting energies, forces, dipole moments, and partial charges[J]. Journal of Chemical Theory and Computation, 2019, 15(6): 3678-3693. |
| 50 | Unke O T, Stöhr M, Ganscha S, et al. Accurate machine learned quantum-mechanical force fields for biomolecular simulations[EB/OL]. 2022. . |
| 51 | Huang B, von Lilienfeld O A. Quantum machine learning using atom-in-molecule-based fragments selected on the fly[J]. Nature Chemistry, 2020, 12: 945-951. |
| 52 | Eckhoff M, Behler J. From molecular fragments to the bulk: development of a neural network potential for MOF-5[J]. Journal of Chemical Theory and Computation, 2019, 15(6): 3793-3809. |
| 53 | Eastman P, Behara P K, Dotson D L, et al. SPICE, a dataset of drug-like molecules and peptides for training machine learning potentials[J]. Scientific Data, 2023, 10: 11. |
| 54 | Pople J A. Nobel lecture: quantum chemical models[J]. Reviews of Modern Physics, 1999, 71(5): 1267-1274. |
| 55 | Bogojeski M, Vogt-Maranto L, Tuckerman M E, et al. Quantum chemical accuracy from density functional approximations via machine learning[J]. Nature Communications, 2020, 11: 5223. |
| 56 | Wang Y S, Wang T, Li S N, et al. Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing[J]. Nature Communications, 2024, 15: 313. |
| 57 | Tokita A M, Behler J. How to train a neural network potential[J]. The Journal of Chemical Physics, 2023, 159(12): 121501. |
| 58 | Chong S, Grasselli F, Ben Mahmoud C, et al. Robustness of local predictions in atomistic machine learning models[J]. Journal of Chemical Theory and Computation, 2023, 19(22): 8020-8031. |
| 59 | Morrow J D, Gardner J L A, Deringer V L. How to validate machine-learned interatomic potentials[J]. The Journal of Chemical Physics, 2023, 158(12): 121501. |
| 60 | Jordan M I, Mitchell T M. Machine learning: trends, perspectives, and prospects[J]. Science, 2015, 349(6245): 255-260. |
| 61 | LeCun Y, Bengio Y, Hinton G. Deep learning[J]. Nature, 2015, 521: 436-444. |
| 62 | Pandey M, Fernandez M, Gentile F, et al. The transformational role of GPU computing and deep learning in drug discovery[J]. Nature Machine Intelligence, 2022, 4: 211-221. |
| 63 | Knight J C, Nowotny T. Larger GPU-accelerated brain simulations with procedural connectivity[J]. Nature Computational Science, 2021, 1: 136-142. |
| 64 | Zhang L F, Han J Q, Wang H, et al. End-to-end symmetry preserving inter-atomic potential energy model for finite and extended systems[C]//Proceedings of the 32nd International Conference on Neural Information Processing Systems. Montréal, Canada: ACM, 2018: 4441-4451. |
| 65 | Zhang L F, Wang H, Car R, et al. Phase diagram of a deep potential water model[J]. Physical Review Letters, 2021, 126(23): 236001. |
| 66 | Zeng J Z, Cao L Q, Xu M Y, et al. Complex reaction processes in combustion unraveled by neural network-based molecular dynamics simulation[J]. Nature Communications, 2020, 11: 5713. |
| 67 | Smith J S, Isayev O, Roitberg A E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost[J]. Chemical Science, 2017, 8(4): 3192-3203. |
| 68 | Gao X, Ramezanghorbani F, Isayev O, et al. TorchANI: a free and open source PyTorch-based deep learning implementation of the ANI neural network potentials[J]. Journal of Chemical Information and Modeling, 2020, 60(7): 3408-3415. |
| 69 | Smith J S, Isayev O, Roitberg A E. ANI-1, a data set of 20 million calculated off-equilibrium conformations for organic molecules[J]. Scientific Data, 2017, 4: 170193. |
| 70 | Yao K, Herr J E, Toth D W, et al. The TensorMol-0.1 model chemistry: a neural network augmented with long-range physics[J]. Chemical Science, 2018, 9(8): 2261-2269. |
| 71 | Desai S, Reeve S T, Belak J F. Implementing a neural network interatomic model with performance portability for emerging exascale architectures[J]. Computer Physics Communications, 2022, 270: 108156. |
| 72 | Kapil V, Schran C, Zen A, et al. The first-principles phase diagram of monolayer nanoconfined water[J]. Nature, 2022, 609: 512-516. |
| 73 | Behler J. Constructing high-dimensional neural network potentials: a tutorial review[J]. International Journal of Quantum Chemistry, 2015, 115(16): 1032-1050. |
| 74 | Behler J. First principles neural network potentials for reactive simulations of large molecular and condensed systems[J]. Angewandte Chemie International Edtion, 2017, 56(42): 12828-12840. |
| 75 | Quaranta V, Hellström M, Behler J. Proton-transfer mechanisms at the water-ZnO interface: the role of presolvation[J]. The Journal of Physical Chemistry Letters, 2017, 8(7): 1476-1483. |
| 76 | Ko T W, Finkler J A, Goedecker S, et al. A fourth-generation high-dimensional neural network potential with accurate electrostatics including non-local charge transfer[J]. Nature Communications, 2021, 12: 398. |
| 77 | Zhang Y L, Hu C, Jiang B. Embedded atom neural network potentials: efficient and accurate machine learning with a physically inspired representation[J]. The Journal of Physical Chemistry Letters, 2019, 10(17): 4962-4967. |
| 78 | Schütt K T, Arbabzadah F, Chmiela S, et al. Quantum-chemical insights from deep tensor neural networks[J]. Nature Communications, 2017, 8: 13890. |
| 79 | Blum L C, Reymond J L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13[J]. Journal of the American Chemical Society, 2009, 131(25): 8732-8733. |
| 80 | Schütt K T, Sauceda H E, Kindermans P J, et al. SchNet — a deep learning architecture for molecules and materials[J]. The Journal of Chemical Physics, 2018, 148(24): 241722. |
| 81 | Schütt K T, Kindermans P J, Sauceda H E, et al. SchNet: a continuous-filter convolutional neural network for modeling quantum interactions[C]//Proceedings of the 31st International Conference on Neural Information Processing Systems. Long Beach, California, USA: ACM, 2017: 992-1002. |
| 82 | Ruddigkeit L, van Deursen R, Blum L C, et al. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17[J]. Journal of Chemical Information and Modeling, 2012, 52(11): 2864-2875. |
| 83 | Ramakrishnan R, Dral P O, Rupp M, et al. Quantum chemistry structures and properties of 134 kilo molecules[J]. Scientific Data, 2014, 1: 140022. |
| 84 | Gasteiger J, Giri S, Margraf J T, et al. Fast and uncertainty-aware directional message passing for non-equilibrium molecules[EB/OL]. 2020. . |
| 85 | Chanussot L, Das A, Goyal S, et al. Open catalyst 2020 (OC20) dataset and community challenges[J]. ACS Catalysis, 2021, 11(10): 6059-6072. |
| 86 | Musaelian A, Batzner S, Johansson A, et al. Learning local equivariant representations for large-scale atomistic dynamics[J]. Nature Communications, 2023, 14: 579. |
| 87 | Batatia I, Kovács D P, Simm G N C, et al. MACE: higher order equivariant message passing neural networks for fast and accurate force fields[EB/OL]. 2022. . |
| 88 | Kovács D P, Oord C V, Kucera J, et al. Linear atomic cluster expansion force fields for organic molecules: beyond RMSE[J]. Journal of Chemical Theory and Computation, 2021, 17(12): 7696-7711. |
| 89 | Lubbers N, Smith J S, Barros K. Hierarchical modeling of molecular energies using a deep neural network[J]. The Journal of Chemical Physics, 2018, 148(24): 241715. |
| 90 | Unke O T, Chmiela S, Gastegger M, et al. SpookyNet: learning force fields with electronic degrees of freedom and nonlocal effects[J]. Nature Communications, 2021, 12: 7273. |
| 91 | Rasmussen C E, Williams C K I. Gaussian Processes for Machine Learning[M]. Cambridge: MIT Press, 2006. |
| 92 | Wood M A, Thompson A P. Extending the accuracy of the SNAP interatomic potential form[J]. The Journal of Chemical Physics, 2018, 148(24): 241721. |
| 93 | Schütt K, Unke O, Gastegger M. Equivariant message passing for the prediction of tensorial properties and molecular spectra[C]//Proceedings of the 38th International Conference on Machine Learning. PMLR, 2021: 9377-9388. |
| 94 | Hofmann T, Schölkopf B, Smola A J. Kernel methods in machine learning[J]. The Annals of Statistics, 2008, 36(3): 1171-1220. |
| 95 | Zhang L F, Wang H, Muniz M C, et al. A deep potential model with long-range electrostatic interactions[J]. The Journal of Chemical Physics, 2022, 156(12): 124107. |
| 96 | Zeng J Z, Giese T J, Ekesan Ş, et al. Development of range-corrected deep learning potentials for fast, accurate quantum mechanical/molecular mechanical simulations of chemical reactions in solution[J]. Journal of Chemical Theory and Computation, 2021, 17(11): 6993-7009. |
| 97 | Zhang D, Bi H R, Dai F Z, et al. DPA-1: pretraining of attention-based deep potential model for molecular simulation[EB/OL]. 2022. . |
| 98 | Zhang D, Liu X, Zhang X Y, et al. DPA-2: towards a universal large atomic model for molecular and material simulation[EB/OL]. 2023. . |
| 99 | Zeng J Z, Zhang D, Lu D H, et al. DeePMD-kit v2: a software package for deep potential models[J]. The Journal of Chemical Physics, 2023, 159(5): 054801. |
| 100 | Smith J S, Nebgen B T, Zubatyuk R, et al. Outsmarting Quantum Chemistry Through Transfer Learning[M/OL]. ChemRxiv, 2018[2024-02-02]. . |
| 101 | Devereux C, Smith J S, Huddleston K K, et al. Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens[J]. Journal of Chemical Theory and Computation, 2020, 16(7): 4192-4202. |
| 102 | Gilmer J, Schoenholz S S, Riley P F, et al. Neural message passing for quantum chemistry[C]//Proceedings of the 34th International Conference on Machine Learning-Volume 70. Sydney, NSW, Australia: ACM, 2017: 1263-1272. |
| 103 | Duval A, Mathis S V, Joshi C K, et al. A hitchhiker's guide to geometric GNNs for 3D atomic systems[EB/OL]. 2023. . |
| 104 | Ghasemi S A, Hofstetter A, Saha S, et al. Interatomic potentials for ionic systems with density functional accuracy based on charge densities obtained by a neural network[J]. Physical Review B, 2015, 92(4): 045131. |
| 105 | Anstine D M, Isayev O. Machine learning interatomic potentials and long-range physics[J]. The Journal of Physical Chemistry. A, 2023, 127(11): 2417-2431. |
| 106 | Jia W, Wang H, Chen M, et al. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning[C]//Proceedings of Sc20: the International Conference for High Performance Computing, Networking, Storage and Analysis (Sc20). New York: IEEE, 2020. |
| 107 | Jose K V, Artrith N, Behler J. Construction of high-dimensional neural network potentials using environment-dependent atom pairs[J]. The Journal of Chemical Physics, 2012, 136(19): 194111. |
| 108 | Artrith N, Behler J. High-dimensional neural network potentials for metal surfaces: a prototype study for copper[J]. Physical Review B, 2012, 85(4): 045439. |
| 109 | Marques M R G, Wolff J, Steigemann C, et al. Neural network force fields for simple metals and semiconductors: construction and application to the calculation of phonons and melting temperatures[J]. Physical Chemistry Chemical Physics, 2019, 21(12): 6506-6516. |
| 110 | Liu J C, Liu R X, Cao Y, et al. Solvation structures of calcium and magnesium ions in water with the presence of hydroxide: a study by deep potential molecular dynamics[J]. Physical Chemistry Chemical Physics: PCCP, 2023, 25(2): 983-993. |
| 111 | Hellström M, Quaranta V, Behler J. One-dimensional vs. two-dimensional proton transport processes at solid-liquid zinc-oxide-water interfaces[J]. Chemical Science, 2018, 10(4): 1232-1243. |
| 112 | Quaranta V, Behler J, Hellström M. Structure and dynamics of the liquid-water/zinc-oxide interface from machine learning potential simulations[J]. The Journal of Physical Chemistry C, 2019, 123(2): 1293-1304. |
| 113 | Natarajan S K, Behler J. Neural network molecular dynamics simulations of solid-liquid interfaces: water at low-index copper surfaces[J]. Physical Chemistry Chemical Physics: PCCP, 2016, 18(41): 28704-28725. |
| 114 | Kozinsky B, Musaelian A, Johansson A, et al. Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size[C]//Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis. Denver, CO, USA: ACM, 2023. |
| 115 | Gallo P, Amann-Winkel K, Angell C A, et al. Water: a tale of two liquids[J]. Chemical Reviews, 2016, 116(13): 7463-7500. |
| 116 | Cisneros G A, Wikfeldt K T, Ojamäe L, et al. Modeling molecular interactions in water: from pairwise to many-body potential energy functions[J]. Chemical Reviews, 2016, 116(13): 7501-7528. |
| 117 | Behler J, Martonák R, Donadio D, et al. Metadynamics simulations of the high-pressure phases of silicon employing a high-dimensional neural network potential[J]. Physical Review Letters, 2008, 100(18): 185501. |
| 118 | Zhang D, Yi P Y, Lai X M, et al. Active machine learning model for the dynamic simulation and growth mechanisms of carbon on metal surface[J]. Nature Communications, 2024, 15: 344. |
| 119 | Gartner T E, Ferguson A L, Debenedetti P G. Data-driven molecular design and simulation in modern chemical engineering[J]. Nature Chemical Engineering, 2024, 1: 6-9. |
| 120 | Kovács D P, Moore J H, Browning N J, et al. MACE-OFF23: transferable machine learning force fields for organic molecules[EB/OL]. 2023. . |
| [1] | Zheng ZHANG, Wuqiong WANG, Yajing ZHANG, Kangjun WANG, Yuanhui JI. Research progress in theoretical calculation of pharmaceutical formulation design [J]. CIESC Journal, 2024, 75(4): 1429-1438. |
| [2] | Kang ZHOU, Jianxin WANG, Hai YU, Chaoliang WEI, Fengqi FAN, Xinhao CHE, Lei ZHANG. Foam rupture properties of mineral base oils based on molecular dynamics simulation [J]. CIESC Journal, 2024, 75(4): 1668-1678. |
| [3] | Haoqi CHEN, Bohui SHI, Qi PENG, Qi KANG, Shangfei SONG, Haiyuan YAO, Haihong CHEN, Haihao WU, Jing GONG. Phase equilibrium calculation of acid/alcohol hydrocarbon and water system based on stability analysis [J]. CIESC Journal, 2024, 75(3): 789-800. |
| [4] | Xinzi ZHOU, Zenghui LI, Xianyang MENG, Jiangtao WU. Experimental study on viscosity of high purity air at low temperatures [J]. CIESC Journal, 2024, 75(3): 782-788. |
| [5] | Fan WU, Xudong PENG, Jinbo JIANG, Xiangkai MENG, Yangyang LIANG. Study on adaptability of molecular dynamics in predicting density and viscosity of natural gas [J]. CIESC Journal, 2024, 75(2): 450-462. |
| [6] | Xin YANG, Wen WANG, Kai XU, Fanhua MA. Simulation analysis of temperature characteristics of the high-pressure hydrogen refueling process [J]. CIESC Journal, 2023, 74(S1): 280-286. |
| [7] | Minghui CHANG, Lin WANG, Jiajia YUAN, Yifei CAO. Study on the cycle performance of salt solution-storage-based heat pump [J]. CIESC Journal, 2023, 74(S1): 329-337. |
| [8] | Minghao SONG, Fei ZHAO, Shuqing LIU, Guoxuan LI, Sheng YANG, Zhigang LEI. Multi-scale simulation and study of volatile phenols removal from simulated oil by ionic liquids [J]. CIESC Journal, 2023, 74(9): 3654-3664. |
| [9] | Jianbo HU, Hongchao LIU, Qi HU, Meiying HUANG, Xianyu SONG, Shuangliang ZHAO. Molecular dynamics simulation insight into translocation behavior of organic cage across the cellular membrane [J]. CIESC Journal, 2023, 74(9): 3756-3765. |
| [10] | Jiajia ZHAO, Shixiang TIAN, Peng LI, Honggao XIE. Microscopic mechanism of SiO2-H2O nanofluids to enhance the wettability of coal dust [J]. CIESC Journal, 2023, 74(9): 3931-3945. |
| [11] | Manzheng ZHANG, Meng XIAO, Peiwei YAN, Zheng MIAO, Jinliang XU, Xianbing JI. Working fluid screening and thermodynamic optimization of hazardous waste incineration coupled organic Rankine cycle system [J]. CIESC Journal, 2023, 74(8): 3502-3512. |
| [12] | Linzheng WANG, Yubing LU, Ruizhi ZHANG, Yonghao LUO. Analysis on thermal oxidation characteristics of VOCs based on molecular dynamics simulation [J]. CIESC Journal, 2023, 74(8): 3242-3255. |
| [13] | Ji CHEN, Ze HONG, Zhao LEI, Qiang LING, Zhigang ZHAO, Chenhui PENG, Ping CUI. Study on coke dissolution loss reaction and its mechanism based on molecular dynamics simulations [J]. CIESC Journal, 2023, 74(7): 2935-2946. |
| [14] | Xiaokun HE, Rui LIU, Yuan XUE, Ran ZUO. Review of gas phase and surface reactions in AlN MOCVD [J]. CIESC Journal, 2023, 74(7): 2800-2813. |
| [15] | Ming DONG, Jinliang XU, Guanglin LIU. Molecular dynamics study on heterogeneous characteristics of supercritical water [J]. CIESC Journal, 2023, 74(7): 2836-2847. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

