CIESC Journal ›› 2020, Vol. 71 ›› Issue (1): 192-199.DOI: 10.11949/0438-1157.20191229
• Thermodynamics • Previous Articles Next Articles
Jie WU( ),Jiahui LI,Yanmei YU,Yangxin YU()
),Jiahui LI,Yanmei YU,Yangxin YU()
Received:2019-09-23
Revised:2019-10-24
Online:2020-01-05
Published:2020-01-05
Contact:
Yangxin YU
吴杰(),李嘉辉,于燕梅,于养信()
通讯作者:
于养信
作者简介:吴杰(1995—),男,博士研究生,基金资助:CLC Number:
Jie WU, Jiahui LI, Yanmei YU, Yangxin YU. Theoretical investigation on thermodynamic properties of group Ⅲ phosphides[J]. CIESC Journal, 2020, 71(1): 192-199.
吴杰, 李嘉辉, 于燕梅, 于养信. 第Ⅲ族元素磷化物热力学性质理论研究[J]. 化工学报, 2020, 71(1): 192-199.
Add to citation manager EndNote|Ris|BibTeX
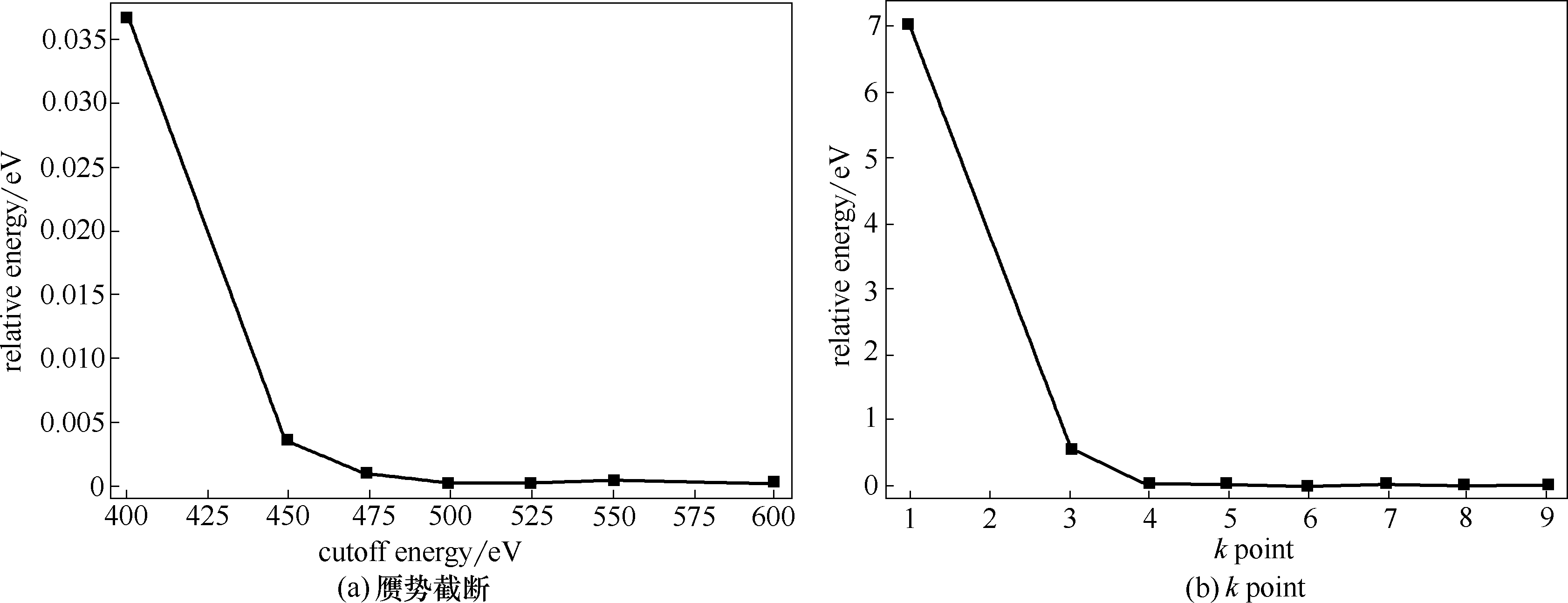
Fig.1 Convergence test
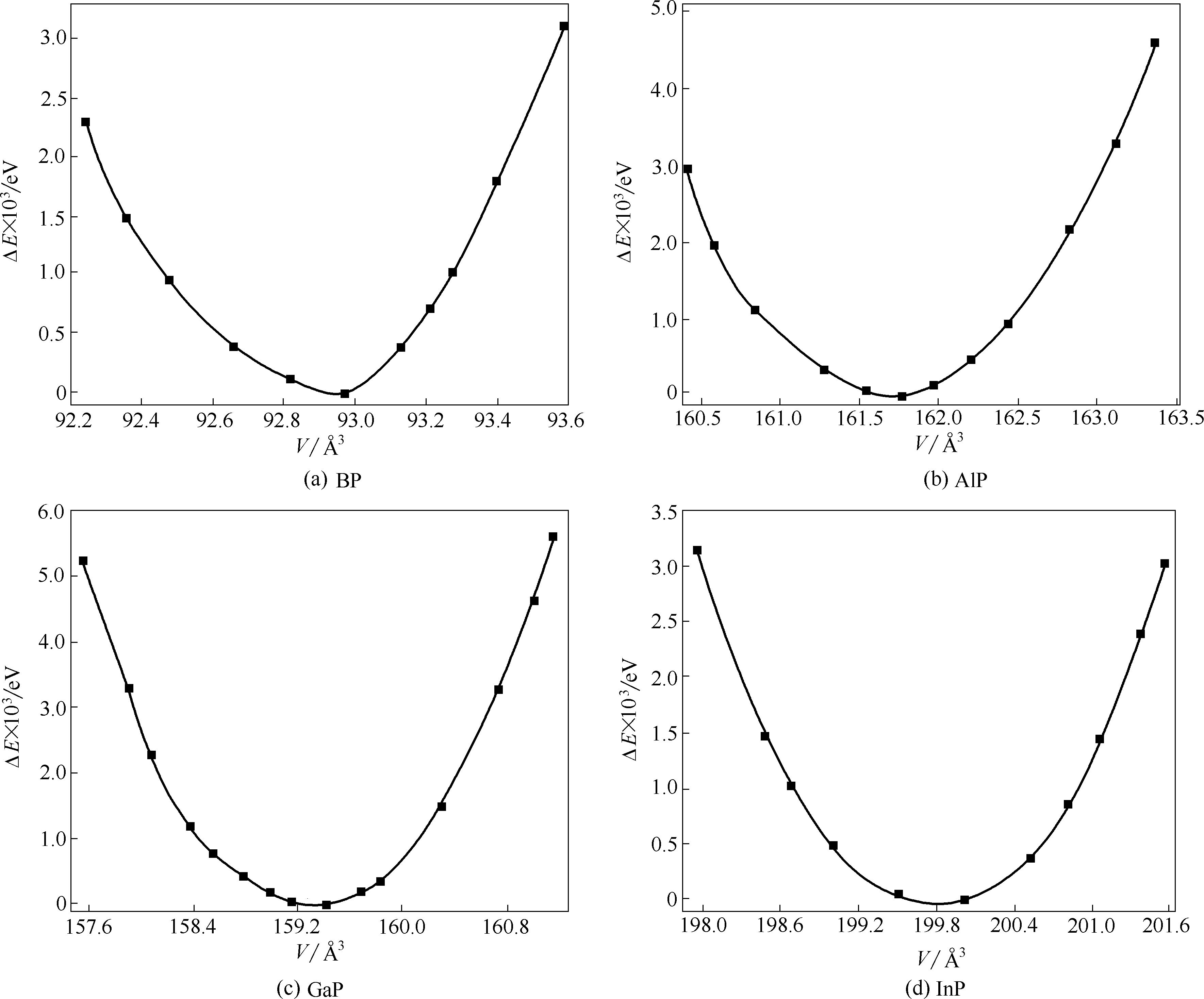
Fig.2 Calculated relative energy (0 K) for group Ⅲ phosphides as a function of volume
| 磷化物 | M-P 键长/ ? | 晶胞边长a/? | 体积模量B/GPa | ||||
|---|---|---|---|---|---|---|---|
| 本文 | 文献 | ||||||
| 0 K | 300 K | 实验(300 K) | 其他计算(0 K) | 本文 | 实验 | ||
| BP AlP GaP InP | 1.96 2.36 2.34 2.53 | 4.530 5.449 5.422 5.848 | 4.531 5.450 5.427 5.850 | 4.54[ 5.45[ 5.45[ 5.87[ | 4.55[ 5.53[ 5.52[ 5.95[ | 161.3 86.7 84.8 64.8 | 173[ 86[ 91[ 72[ |
Table 1 Bond lengths, lattice constants a and bulk modulus for zinc-blende metal phosphides
| 磷化物 | M-P 键长/ ? | 晶胞边长a/? | 体积模量B/GPa | ||||
|---|---|---|---|---|---|---|---|
| 本文 | 文献 | ||||||
| 0 K | 300 K | 实验(300 K) | 其他计算(0 K) | 本文 | 实验 | ||
| BP AlP GaP InP | 1.96 2.36 2.34 2.53 | 4.530 5.449 5.422 5.848 | 4.531 5.450 5.427 5.850 | 4.54[ 5.45[ 5.45[ 5.87[ | 4.55[ 5.53[ 5.52[ 5.95[ | 161.3 86.7 84.8 64.8 | 173[ 86[ 91[ 72[ |
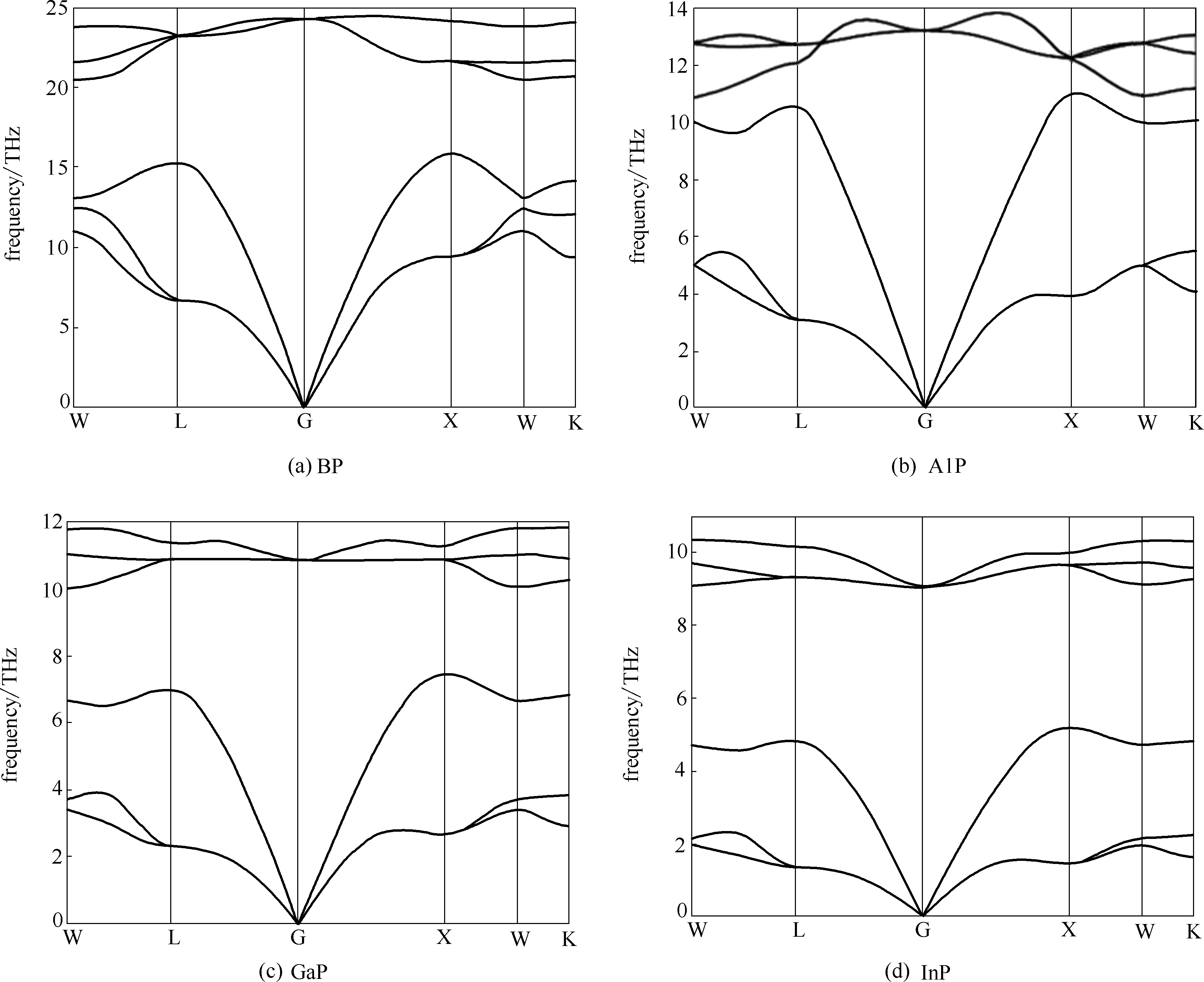
Fig.3 Calculated phonon dispersion curves for group Ⅲ phosphides at 0 K
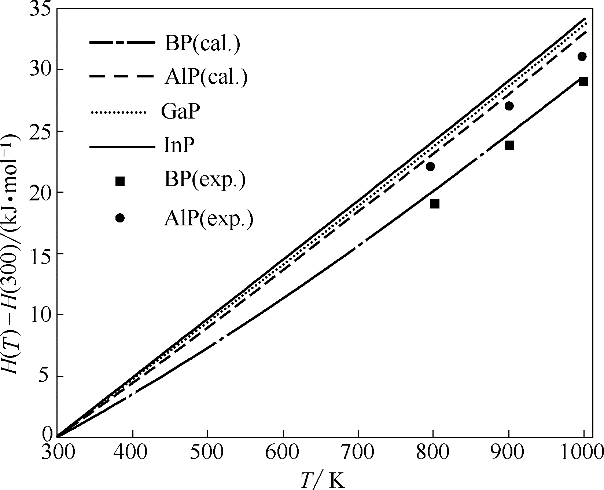
Fig.4 Calculated enthalpy as a function of temperature for BP, AlP, GaP and InP (H(300) is enthalpy at 300 K)
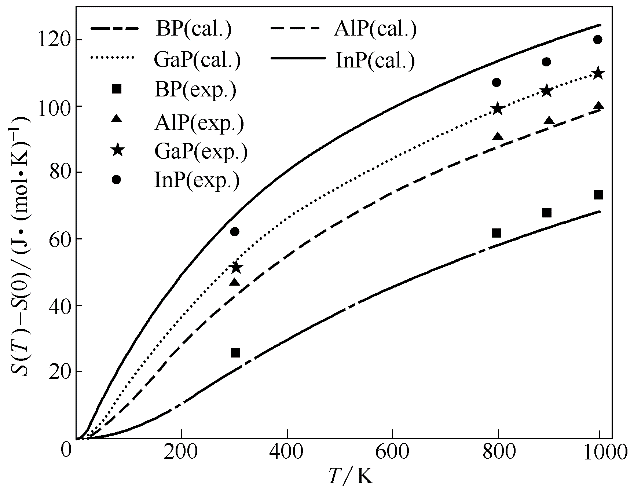
Fig.5 Calculated entropy as a function of temperature for BP, AlP, GaP and InP (S(0) is entropy at 0 K)
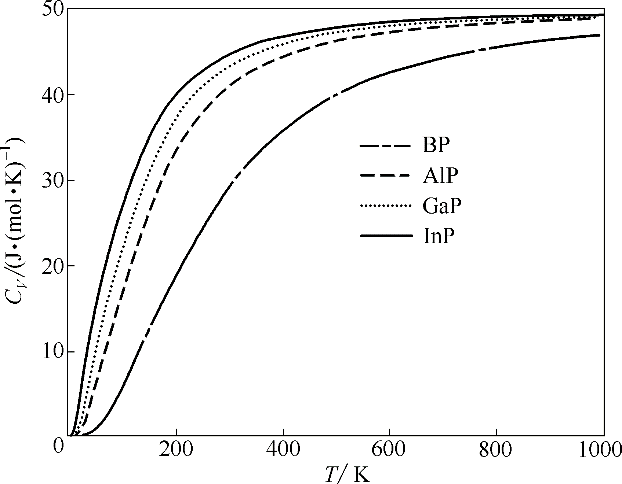
Fig.6 Calculated heat capacity as a function of temperature for BP, AlP, GaP and InP
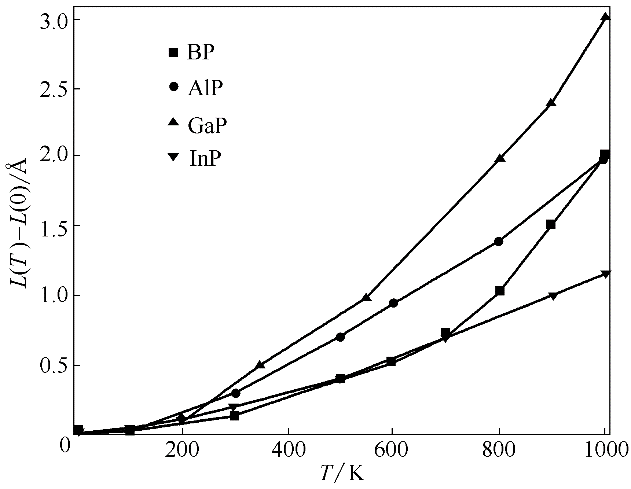
Fig.7 Calculated equilibrium lattice parameter as a function of temperature for BP, AlP, GaP and InP (L(0) is equilibrium lattice parameter at 0 K)
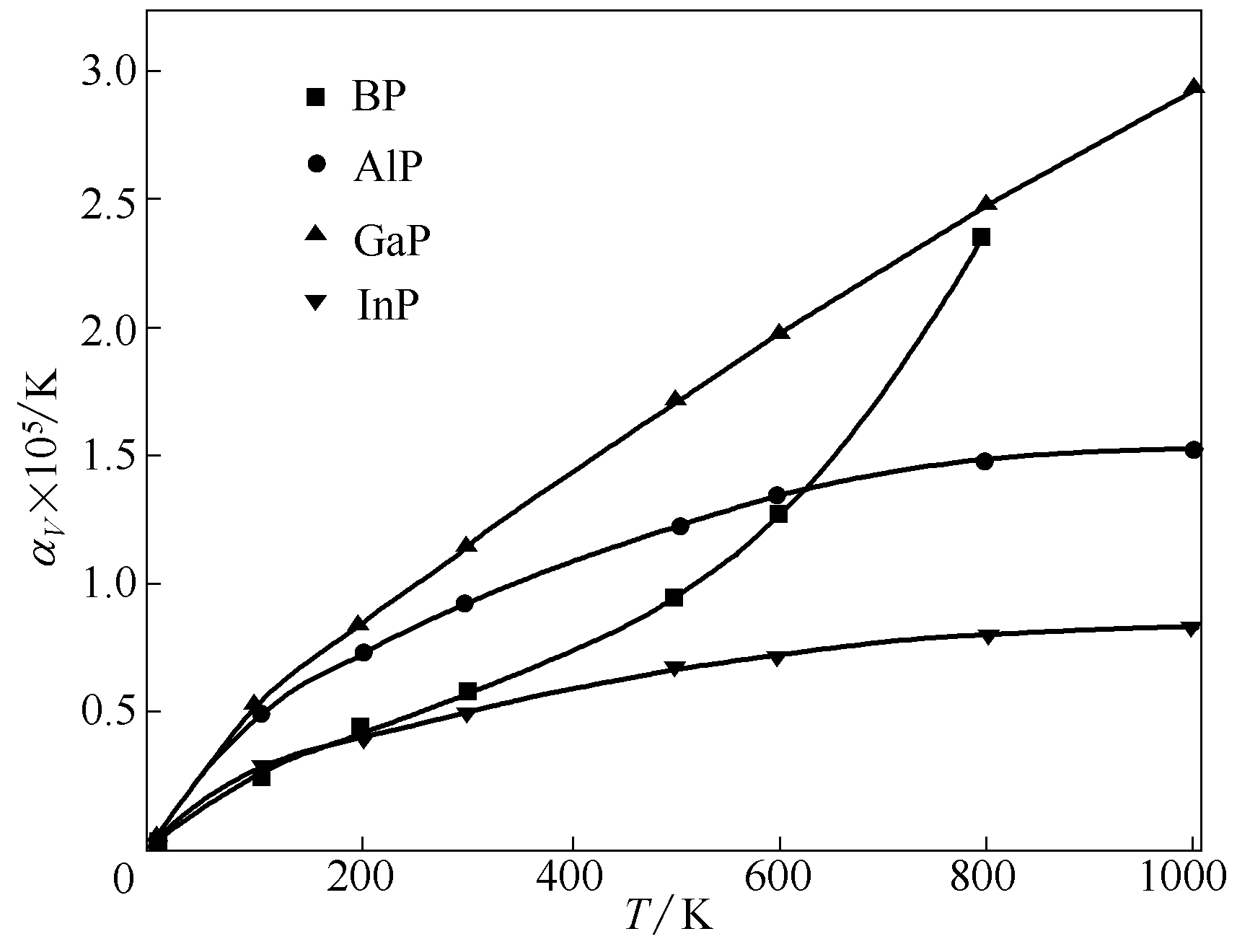
Fig.8 Calculated volume thermal-expansion coefficient as a function of temperature for BP, AlP, GaP and InP
| 1 | Vurgaftman I , Meyer J R , Ram-Mohan L R . Band parameters for Ⅲ-V compound semiconductors and their alloys[J]. Journal of Applied Physics, 2001, 89(11): 5815-5875. |
| 2 | Yu L H , Yao K L , Liu Z L . Electronic band structures of filled tetrahedral semiconductor LiMgP and zinc-blende AlP[J]. Solid State Communications, 2005, 135(1/2): 124-128. |
| 3 | Zaoui A , Kacimi S , Yakoubi A , et al . Optical properties of BP, BAs and BSb compounds under hydrostatic pressure[J]. Physica B-Condensed Matter, 2005, 367(1/2/3/4): 195-204. |
| 4 | Touat D , Ferhat M , Zaoui A . Dynamical behaviour in the boron Ⅲ-Ⅴ group: a first-principles study[J]. Journal of Physics-Condensed Matter, 2006, 18(15): 3647-3654. |
| 5 | Lu X F , Gao X , Li C X , et al . Electronic structure and optical properties of doped gallium phosphide: a first-principles simulation[J]. Physics Letters A, 2017, 381(35): 2986-2992. |
| 6 | Tong C J , Zhang H , Zhang Y N , et al . New manifold two-dimensional single-layer structures of zinc-blende compounds[J]. Journal of Materials Chemistry A, 2014, 2(42): 17971-17978. |
| 7 | Fornari R . Single Crystals of Electronic Materials: Growth and Properties[M]. Cambridge: Woodhead Publ. Ltd., 2019: 241-268. |
| 8 | Crisp R W , Kirkwood N , Grimaldi G , et al . Highly photoconductive InP quantum dots films and solar cells[J]. ACS Applied Energy Materials, 2018, 1(11): 6569-6576. |
| 9 | Li S , Taddei K M , Wang X Q , et al . Thermal expansion coefficients of high thermal conducting BAs and BP materials[J]. Applied Physics Letters, 2019, 115(1): 011901. |
| 10 | Abrishamifar S M , Heidari N , Razavi R , et al . The Cl functionalized aluminum nitride (AlN) and aluminum phosphide (AlP) nanocone sheets as hydrogen selenide (H2Se) sensor: a density functional investigation[J]. Acta Chimica Slovenica, 2018, 65(1): 208-212. |
| 11 | Chen Z H , Shao Z H , Siddiqui M K , et al . Potential of carbon, silicon, boron nitride and aluminum phosphide nanocages as anodes of lithium, sodium and potassium ion batteries: a DFT study[J]. Russian Journal of Physical Chemistry B, 2019, 13(1): 156-164. |
| 12 | Halmann M . Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells[J]. Nature, 1978, 275(5676): 115-116. |
| 13 | Jiao Z Y , Ma S H , Guo Y L . Simulation of optical function for phosphide crystals following the DFT band structure calculations[J]. Computational and Theoretical Chemistry, 2011, 970(1/2/3): 79-84. |
| 14 | Britto R J , Young J L , Yang Y , et al . Interfacial engineering of gallium indium phosphide photoelectrodes for hydrogen evolution with precious metal and non-precious metal based catalysts[J]. Journal of Materials Chemistry A, 2019, 7(28): 16821-16832. |
| 15 | Hestroffer K , Sperlich D , Dadgostar S , et al . Transport properties of doped AlP for the development of conductive AlP/GaP distributed Bragg reflectors and their integration into light-emitting diodes[J]. Applied Physics Letters, 2018, 112(19): 191207. |
| 16 | Zhao S , Butera S , Lioliou G , et al . AlInP photodiode X-ray detectors[J]. Journal of Physics D-Applied Physics, 2019, 52(22): 225101. |
| 17 | Mujica A , Rubio A , Munoz A , et al . High-pressure phases of group-IV, Ⅲ-V, and II-VI compounds[J]. Reviews of Modern Physics, 2003, 75(3): 863-912. |
| 18 | Saib S , Bouarissa N , Rodriguez-Hernandez P , et al . Elastic modulus and thermal properties of InN in the rocksalt phase[J]. Computational Materials Science, 2014, 81: 374-377. |
| 19 | Bioud N , Kassali K , Bouarissa N . Thermodynamic properties of compressed CuX (X = Cl, Br) compounds: ab initio study[J]. Journal of Electronic Materials, 2017, 46(4): 2521-2528. |
| 20 | Daoud S , Bouarissa N . Structural and thermodynamic properties of cubic sphalerite aluminum nitride under hydrostatic compression[J]. Computational Condensed Matter, 2019, 19: e00359. |
| 21 | Daoud S , Bouarissa N , Bioud N , et al . High-temperature and high-pressure thermophysical properties of AlP semiconducting material: a systematic ab initio study[J]. Chemical Physics, 2019, 525: UNSP 110399. |
| 22 | Settouf A , Rached H , Benkhettou N , et al . DFT calculations of structural, optoelectronic and thermodynamic properties of B x Al1 -x P alloys[J]. Computational Condensed Matter, 2019, 19: e00377. |
| 23 | Segall M D , Lindan P J D , Probert M J , et al . First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics-Condensed Matter, 2002, 14(11): 2717-2744. |
| 24 | Lejaeghere K , Bihlmayer G , Björkman T , et al . Reproducibility in density functional theory calculations of solids[J]. Science, 2016, 351(6280): aad3000. |
| 25 | Perdew J P , Burke K , Ernzerhof M . Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868. |
| 26 | Grimme S . Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J]. Journal of Computational Chemistry, 2006, 27(15): 1787-1799. |
| 27 | Baroni S , de Gironcoli S , Dal Corso A , et al . Phonons and related crystal properties from density-functional perturbation theory[J]. Reviews of Modern Physics, 2001, 73(2): 515-562. |
| 28 | Wyckoff R W G . Crystal Structures[M]. 2nd ed. New York: John Wiley & Sons Inc., 1964: 110-112. |
| 29 | Xia H , Xia Q , Ruoff A L . BP at megabar pressures and its equation of state to 110 GPa[J]. Journal of Applied Physics, 1993, 74(3): 1660-1662. |
| 30 | Lambrecht W R L , Segall B . Electronic-structure and bonding at SiC/AlN and SiC/BP interfaces[J]. Physical Review B, 1991, 43(9): 7070-7085. |
| 31 | Kalvoda S , Paulus B , Fulde P , et al . Influence of electron correlations on ground-state properties of Ⅲ-V semiconductors[J]. Physical Review B, 1997, 55(7): 4027-4030. |
| 32 | Wang S Q , Ye H Q . A plane-wave pseudopotential study on Ⅲ-V zinc-blende and wurtzite semiconductors under pressure[J]. Journal of Physics-Condensed Matter, 2002, 14(41): 9579-9587. |
| 33 | Zafar M , Masood M K , Rizwan M , et al . Theoretical study of structural, electronic, optical and elastic properties of Al x Ga 1-x P[J]. Optik, 2019, 182: 1176-1185. |
| 34 | Ahmed R , Fazal E A , Hashemifar S J , et al . First-principles study of the structural and electronic properties of Ⅲ-phosphides[J]. Physica B-Condensed Matter, 2008, 403(10/11): 1876-1881. |
| 35 | Vancamp P E , Vandoren V E , Devreese J T . Pressure-dependence of the electronic-properties of cubic Ⅲ-V in compounds[J]. Physical Review B, 1990, 41(3): 1598-1602. |
| 36 | Schroten E , Goossens A , Schoonman J . Photo- and electroreflectance of cubic boron phosphide[J]. Journal of Applied Physics, 1998, 83(3): 1660-1663. |
| 37 | Madelung O . Semiconductors: Data Handbook[M]. Berlin: Springer, 2004. |
| 38 | Lu Y L , Jia D W , Gao F , et al . First-principle calculations of the thermal properties of SrTiO3 and SrO(SrTiO3) n (n=1,2)[J]. Solid State Communications, 2015, 201: 25-30. |
| 39 | Dumont H , Montell Y . Some aspects on thermodynamic properties, phase diagram and alloy formation in the ternary system BAs-GaAs(Ⅰ): Analysis of BAs thermodynamic properties[J]. Journal of Crystal Growth, 2006, 290(2): 410-418. |
| 40 | Hou B S , Liu K , Mao X C , et al . Theoretical calculations for elastic and thermodynamic properties of NbN2 under high pressure[J]. Acta Physica Polonica A, 2017, 132(4): 1363-1370. |
| 41 | Lebga N , Daoud S , Sun X W , et al . Mechanical and thermophysical properties of cubic rock-salt AlN under high pressure[J]. Journal of Electronic Materials, 2018, 47(7): 3430-3439. |
| 42 | Ö Çiftci Y , Çolakoğlu K , Deligöz E , et al . First-principles calculations on structure, elastic and thermodynamic properties of Al2X (X=Sc, Y) under pressure[J]. Journal of Materials Science & Technology, 2012, 28(2): 155-163. |
| 43 | Xing M , Li B , Yu Z , et al . Elastic anisotropic and thermodynamic properties of I-4m2-BCN[J]. Acta Physica Polonica A, 2016, 129(6): 1124-1130. |
| 44 | Bouhafs B , Aourag H , Certier M . Trends in band-gap pressure coefficients in boron compounds BP, BAs, and BSb[J]. Journal of Physics: Condensed Matter, 2000, 12(26): 5655-5668. |
| [1] | Guang WANG, Fashun SHAN, Yucheng QIAN, Jianfang JIAO. Incipient fault detection method for chemical process based on ensemble learning transfer entropy [J]. CIESC Journal, 2023, 74(7): 2967-2978. |
| [2] | Jinyu GUO, Zhe WANG, Yuan LI. Fault detection method based on kernel entropy independent component analysis [J]. CIESC Journal, 2022, 73(8): 3647-3658. |
| [3] | Zhe SUN, Huaqiang JIN, Kang LI, Jiangping GU, Yuejin HUANG, Xi SHEN. Fault diagnosis method of refrigeration and air-conditioning system based on digitized knowledge representation [J]. CIESC Journal, 2022, 73(7): 3131-3144. |
| [4] | Wenxin MEN, Qingshou PENG, Xia GUI. Phase equilibrium of CO2 hydrate in the presence of four different quaternary ammonium salts [J]. CIESC Journal, 2022, 73(4): 1472-1482. |
| [5] | Hao XU, Wei CHEN, Zoulu LI. Study on the characteristics of the second type heat pump with [Li(TX-7)]SCN/H2O as the working fluid pair [J]. CIESC Journal, 2022, 73(2): 577-586. |
| [6] | Pengpeng WANG, Yanggang JIA, Xia SHAO, Jie CHENG, Aiqin MAO, Jie TAN, Daolai FANG. Preparation and lithium storage performance of K+-doped spinel (Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4 high-entropy oxide anode materials [J]. CIESC Journal, 2022, 73(12): 5625-5637. |
| [7] | CUI Yunhao, QIAO Jianxin, WANG Xiaotao, SONG Bin, YANG Zhaohui, DAI Wei, LI Haibing. Stirling cooler operating in room temperature [J]. CIESC Journal, 2021, 72(S1): 390-397. |
| [8] | Zhirong CHEN, Yun TONG, Shenfeng YUAN, Hong YIN. Dissociation constants and activity coefficients of methionine in KCl aqueous solutions [J]. CIESC Journal, 2021, 72(9): 4469-4478. |
| [9] | Jinyu GUO, Wentao LI, Yuan LI. Application of adaptive algorithm of online reduced KECA in fault detection [J]. CIESC Journal, 2021, 72(8): 4227-4238. |
| [10] | SONG Bennan, WU Chunmei, LI Yourong. Investigation on cluster distribution and phase transition of adsorption at solid-vapor interface [J]. CIESC Journal, 2021, 72(5): 2680-2687. |
| [11] | CHEN Xili, SUN Guoming, JIA Shengkun, LUO Yiqing, YUAN Xigang. Identification of rules for optimal synthesis of ternary-distillation configuration based on decision tree [J]. CIESC Journal, 2021, 72(3): 1430-1437. |
| [12] | Dong WANG, Yaru LIU, Zhuo CHEN, Zunli KOU, Yuehong LU. Effects on performance of small water-source heat pump water heater with CO2 by refrigerant charge and determination of optimal value [J]. CIESC Journal, 2020, 71(S1): 397-403. |
| [13] | Jiannian HAN, Gang WANG, Mei YANG, Meijia LIU, Chengdi GAO, Jinsen GAO. Thermodynamic study on fluid catalytic cracking of Fischer-Tropsch wax to produce clean gasoline [J]. CIESC Journal, 2020, 71(S1): 38-45. |
| [14] | Tingwei ZHANG, Bin LI, Xiaoqiang ZHAI. Topology optimization on heat conduction based on entransy theory [J]. CIESC Journal, 2020, 71(S1): 31-37. |
| [15] | Jianpei CHANG, Xiang HUANG, Miaomiao AN, Zhaoyang LI. Analysis of principle, performance and applicability of indirect evaporative water chiller [J]. CIESC Journal, 2020, 71(S1): 236-244. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

