CIESC Journal ›› 2024, Vol. 75 ›› Issue (11): 4141-4151.DOI: 10.11949/0438-1157.20240602
• Thermodynamics • Previous Articles Next Articles
Yewei DING( ), Wenbo KANG, Yutong SONG, Qinxi FAN, Yuanhui JI()
), Wenbo KANG, Yutong SONG, Qinxi FAN, Yuanhui JI()
Received:2024-06-03
Revised:2024-10-01
Online:2024-12-26
Published:2024-11-25
Contact:
Yuanhui JI
丁叶薇(), 康文博, 宋昱潼, 樊钦习, 吉远辉()
通讯作者:
吉远辉
作者简介:丁叶薇(2000—),女,硕士研究生,dingyewei@seu.edu.cn
基金资助:CLC Number:
Yewei DING, Wenbo KANG, Yutong SONG, Qinxi FAN, Yuanhui JI. Mechanism and screening of indomethacin self-assembled nanomedical drugs[J]. CIESC Journal, 2024, 75(11): 4141-4151.
丁叶薇, 康文博, 宋昱潼, 樊钦习, 吉远辉. 吲哚美辛纳米药物筛选及自组装机制的理论研究[J]. 化工学报, 2024, 75(11): 4141-4151.
Add to citation manager EndNote|Ris|BibTeX
| 类别 | 药物名称及缩写 | SMILES结构编码 | ||
|---|---|---|---|---|
| 非甾体抗炎药 | 吲哚美辛 | Indomethacin | IND | CC1=C(C2=C(N1C(=O)C3=CC=C(C=C3)Cl)C=CC(=C2)OC)CC(=O)O |
| 抗代谢类药物 | 氟达拉滨 | Fludarabine | FD | C1=NC2=C(N=C(N=C2N1C3C(C(C(O3)CO)O)O)F)N |
| 卡培他滨 | Capecitabine | CA | CCCCCOC(=O)NC1=NC(=O)N(C=C1F)[C@@H]1O[C@H](C)[C@@H](O)[C@H]1O | |
| 抗生素类 | 柔红霉素 | Daunorubicin | DA | COC1=CC=CC2=C1C(=O)C1=C(O)C3=C(C[C@](O)(C[C@@H]3O[C@H]3C[C@H](N)[C@H](O)[C@H](C)O3)C(C)=O)C(O)=C1C2=O |
| 阿霉素 | Adriamycin | AD | COC1=CC=CC2=C1C(=O)C1=C(O)C3=C(C[C@](O)(C[C@@H]3O[C@H]3C[C@H](N)[C@H](O)[C@H](C)O3)C(=O)CO)C(O)=C1C2=O | |
| 拓扑异构酶类 | 伊立替康 | Irinotecan | IR | CCC1=C2CN3C(=CC4=C(COC(=O)[C@]4(O)CC)C3=O)C2=NC2=CC=C(OC(=O)N3CCC(CC3)N3CCCCC3)C=C12 |
| 拓扑替康 | Topotecan | TP | CC[C@@]1(C2=C(COC1=O)C(=O)N3CC4=CC5=C(C=CC(=C5CN(C)C)O)N=C4C3=C2)O | |
| 激素类 | 他莫昔芬 | Tamoxifen | TF | CC/C(=C(\C1=CC=CC=C1)/C2=CC=C(C=C2)OCCN(C)C)/C3=CC=CC=C3 |
| 阿那曲唑 | Anastrozole | AZ | CC(C)(C#N)C1=CC(=CC(=C1)CN2C=NC=N2)C(C)(C)C#N | |
| 依西美坦 | Exemestane | ET | C[C@]12CC[C@H]3[C@H]([C@@H]1CCC2=O)CC(=C)C4=CC(=O)C=C[C@]34C | |
| 血管内皮生长 因子受体类 | 帕唑帕尼 | Pazopanib | PB | CC1=C(C=C(C=C1)NC2=NC=CC(=N2)N(C)C3=CC4=NN(C(=C4C=C3)C)C)S(=O)(=O)N |
| 阿西替尼 | Axitinib | AX | CNC(=O)C1=CC=CC=C1SC2=CC3=C(C=C2)C(=NN3)/C=C/C4=CC=CC=N4 | |
| 蛋白酶体 抑制剂类 | 硼替佐米 | Bortezomib | BZ | B([C@H](CC(C)C)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2=NC=CN=C2)(O)O |
| 转谷氨酰胺酶2 (TGase2)抑制剂 | GK921 | GK | C1CCN(C1)CCOC2=NC3=C(N=CC=C3)N=C2C#CC4=CC=CC=C4 | |
| 组蛋白去乙酰化酶类 | 贝利司他 | Belinostat | BE | ONC(=O)\C=C\C1=CC=CC(=C1)S(=O)(=O)NC1=CC=CC=C1 |
Table 1 The drugs selected in this paper
| 类别 | 药物名称及缩写 | SMILES结构编码 | ||
|---|---|---|---|---|
| 非甾体抗炎药 | 吲哚美辛 | Indomethacin | IND | CC1=C(C2=C(N1C(=O)C3=CC=C(C=C3)Cl)C=CC(=C2)OC)CC(=O)O |
| 抗代谢类药物 | 氟达拉滨 | Fludarabine | FD | C1=NC2=C(N=C(N=C2N1C3C(C(C(O3)CO)O)O)F)N |
| 卡培他滨 | Capecitabine | CA | CCCCCOC(=O)NC1=NC(=O)N(C=C1F)[C@@H]1O[C@H](C)[C@@H](O)[C@H]1O | |
| 抗生素类 | 柔红霉素 | Daunorubicin | DA | COC1=CC=CC2=C1C(=O)C1=C(O)C3=C(C[C@](O)(C[C@@H]3O[C@H]3C[C@H](N)[C@H](O)[C@H](C)O3)C(C)=O)C(O)=C1C2=O |
| 阿霉素 | Adriamycin | AD | COC1=CC=CC2=C1C(=O)C1=C(O)C3=C(C[C@](O)(C[C@@H]3O[C@H]3C[C@H](N)[C@H](O)[C@H](C)O3)C(=O)CO)C(O)=C1C2=O | |
| 拓扑异构酶类 | 伊立替康 | Irinotecan | IR | CCC1=C2CN3C(=CC4=C(COC(=O)[C@]4(O)CC)C3=O)C2=NC2=CC=C(OC(=O)N3CCC(CC3)N3CCCCC3)C=C12 |
| 拓扑替康 | Topotecan | TP | CC[C@@]1(C2=C(COC1=O)C(=O)N3CC4=CC5=C(C=CC(=C5CN(C)C)O)N=C4C3=C2)O | |
| 激素类 | 他莫昔芬 | Tamoxifen | TF | CC/C(=C(\C1=CC=CC=C1)/C2=CC=C(C=C2)OCCN(C)C)/C3=CC=CC=C3 |
| 阿那曲唑 | Anastrozole | AZ | CC(C)(C#N)C1=CC(=CC(=C1)CN2C=NC=N2)C(C)(C)C#N | |
| 依西美坦 | Exemestane | ET | C[C@]12CC[C@H]3[C@H]([C@@H]1CCC2=O)CC(=C)C4=CC(=O)C=C[C@]34C | |
| 血管内皮生长 因子受体类 | 帕唑帕尼 | Pazopanib | PB | CC1=C(C=C(C=C1)NC2=NC=CC(=N2)N(C)C3=CC4=NN(C(=C4C=C3)C)C)S(=O)(=O)N |
| 阿西替尼 | Axitinib | AX | CNC(=O)C1=CC=CC=C1SC2=CC3=C(C=C2)C(=NN3)/C=C/C4=CC=CC=N4 | |
| 蛋白酶体 抑制剂类 | 硼替佐米 | Bortezomib | BZ | B([C@H](CC(C)C)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2=NC=CN=C2)(O)O |
| 转谷氨酰胺酶2 (TGase2)抑制剂 | GK921 | GK | C1CCN(C1)CCOC2=NC3=C(N=CC=C3)N=C2C#CC4=CC=CC=C4 | |
| 组蛋白去乙酰化酶类 | 贝利司他 | Belinostat | BE | ONC(=O)\C=C\C1=CC=CC(=C1)S(=O)(=O)NC1=CC=CC=C1 |
| δd | δp | δh | δ | Δδ | δV | Δδd | Δδp | Δδh | Ra | |
|---|---|---|---|---|---|---|---|---|---|---|
| IND | 20.7 | 6.5 | 6.8 | 22.7 | 0 | 21.70 | 0 | 0 | 0 | 0 |
| FD | 18.7 | 14.4 | 16.4 | 28.8 | 6.1 | 23.60 | 2.0 | 7.9 | 9.6 | 14.78 |
| CA | 17.4 | 10.5 | 8.5 | 22.0 | 0.7 | 20.32 | 3.3 | 4.0 | 1.7 | 13.90 |
| DA | 20.7 | 12.6 | 7.8 | 25.5 | 2.8 | 24.23 | 0 | 6.1 | 1.0 | 6.18 |
| AD | 20.8 | 13.6 | 7.4 | 25.9 | 3.2 | 24.85 | 0.1 | 7.1 | 0.6 | 7.14 |
| IR | 20.7 | 4.1 | 7.7 | 22.4 | 0.3 | 21.10 | 0 | 2.4 | 0.9 | 2.56 |
| TP | 20.0 | 3.1 | 10.8 | 23.0 | 0.3 | 20.24 | 0.7 | 3.4 | 4.0 | 5.95 |
| TF | 18.4 | 2.9 | 3.6 | 19.0 | 3.7 | 18.63 | 2.3 | 3.6 | 3.2 | 10.38 |
| AZ | 18.0 | 11.1 | 3.8 | 21.5 | 1.2 | 21.15 | 2.7 | 4.6 | 3.0 | 12.12 |
| ET | 18.9 | 5.4 | 2.8 | 19.9 | 2.8 | 19.66 | 1.8 | 1.1 | 4.0 | 8.31 |
| PB | 21.5 | 10.4 | 7.9 | 25.1 | 2.4 | 23.88 | 0.8 | 3.9 | 1.1 | 5.16 |
| AX | 22.4 | 11.1 | 6.2 | 25.8 | 3.1 | 25.00 | 1.7 | 4.6 | 0.6 | 8.23 |
| BZ | 19.4 | 11.0 | 18.5 | 29.0 | 6.3 | 22.30 | 1.3 | 4.5 | 11.7 | 13.57 |
| GK | 19.1 | 6.6 | 4.8 | 20.8 | 1.9 | 20.21 | 1.6 | 0.1 | 2.0 | 6.71 |
| BE | 20.9 | 14.9 | 15.9 | 30.2 | 7.5 | 25.67 | 0.2 | 8.4 | 9.1 | 12.41 |
Table 2 Hansen solubility parameters
| δd | δp | δh | δ | Δδ | δV | Δδd | Δδp | Δδh | Ra | |
|---|---|---|---|---|---|---|---|---|---|---|
| IND | 20.7 | 6.5 | 6.8 | 22.7 | 0 | 21.70 | 0 | 0 | 0 | 0 |
| FD | 18.7 | 14.4 | 16.4 | 28.8 | 6.1 | 23.60 | 2.0 | 7.9 | 9.6 | 14.78 |
| CA | 17.4 | 10.5 | 8.5 | 22.0 | 0.7 | 20.32 | 3.3 | 4.0 | 1.7 | 13.90 |
| DA | 20.7 | 12.6 | 7.8 | 25.5 | 2.8 | 24.23 | 0 | 6.1 | 1.0 | 6.18 |
| AD | 20.8 | 13.6 | 7.4 | 25.9 | 3.2 | 24.85 | 0.1 | 7.1 | 0.6 | 7.14 |
| IR | 20.7 | 4.1 | 7.7 | 22.4 | 0.3 | 21.10 | 0 | 2.4 | 0.9 | 2.56 |
| TP | 20.0 | 3.1 | 10.8 | 23.0 | 0.3 | 20.24 | 0.7 | 3.4 | 4.0 | 5.95 |
| TF | 18.4 | 2.9 | 3.6 | 19.0 | 3.7 | 18.63 | 2.3 | 3.6 | 3.2 | 10.38 |
| AZ | 18.0 | 11.1 | 3.8 | 21.5 | 1.2 | 21.15 | 2.7 | 4.6 | 3.0 | 12.12 |
| ET | 18.9 | 5.4 | 2.8 | 19.9 | 2.8 | 19.66 | 1.8 | 1.1 | 4.0 | 8.31 |
| PB | 21.5 | 10.4 | 7.9 | 25.1 | 2.4 | 23.88 | 0.8 | 3.9 | 1.1 | 5.16 |
| AX | 22.4 | 11.1 | 6.2 | 25.8 | 3.1 | 25.00 | 1.7 | 4.6 | 0.6 | 8.23 |
| BZ | 19.4 | 11.0 | 18.5 | 29.0 | 6.3 | 22.30 | 1.3 | 4.5 | 11.7 | 13.57 |
| GK | 19.1 | 6.6 | 4.8 | 20.8 | 1.9 | 20.21 | 1.6 | 0.1 | 2.0 | 6.71 |
| BE | 20.9 | 14.9 | 15.9 | 30.2 | 7.5 | 25.67 | 0.2 | 8.4 | 9.1 | 12.41 |
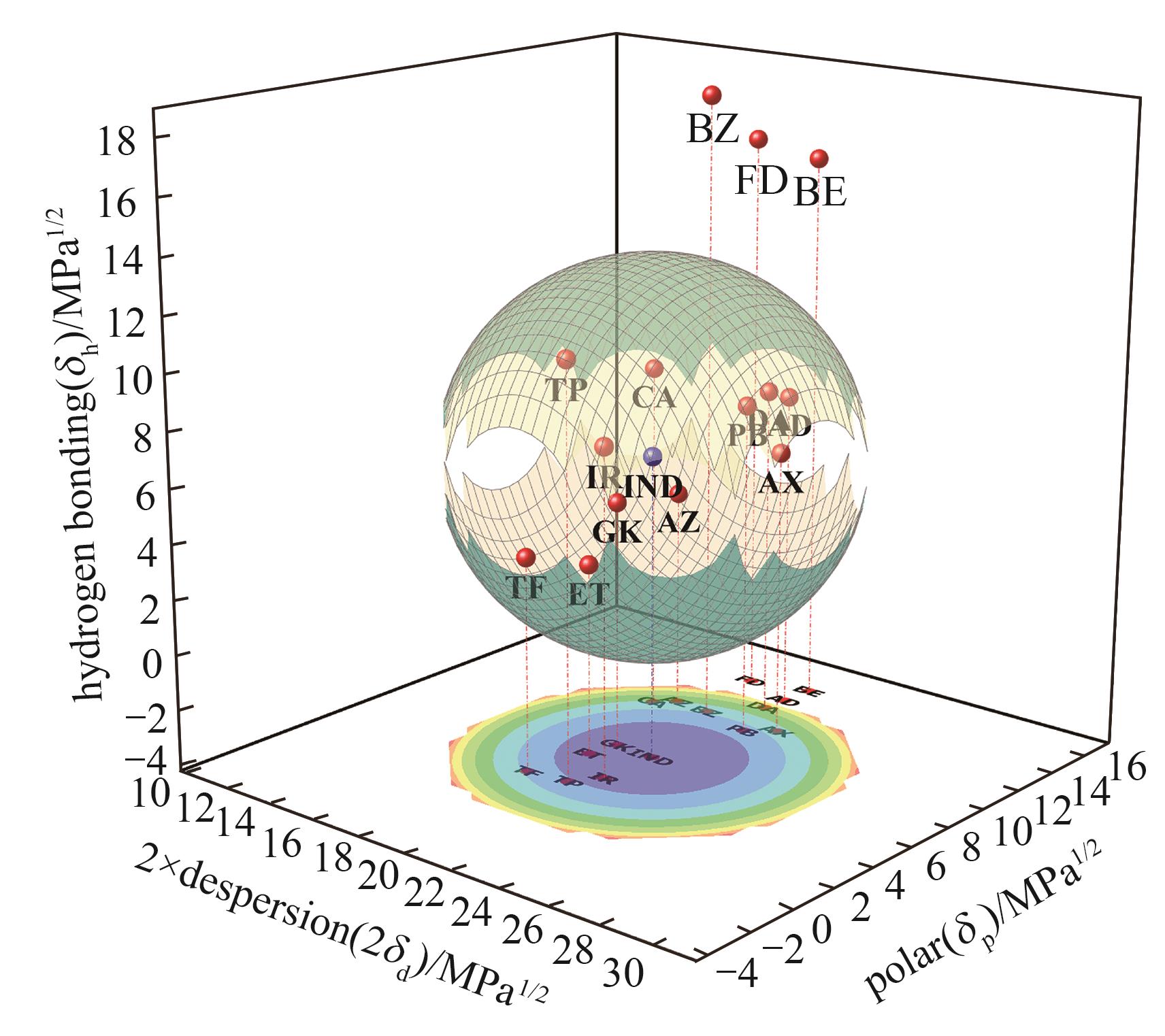
Fig.1 Hansen solubility parameters sphere
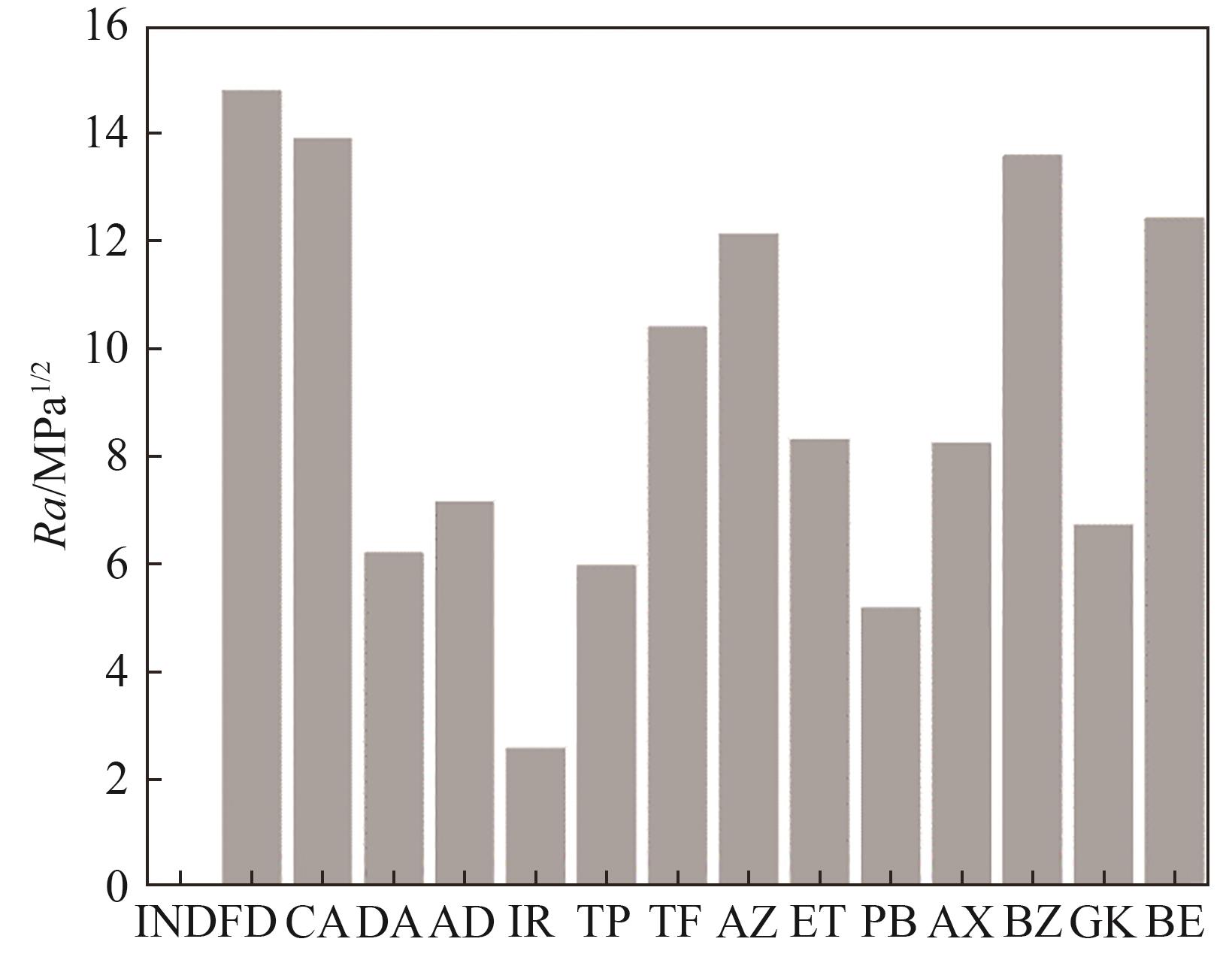
Fig.2 Modified radius (Ra) of the candidate drugs
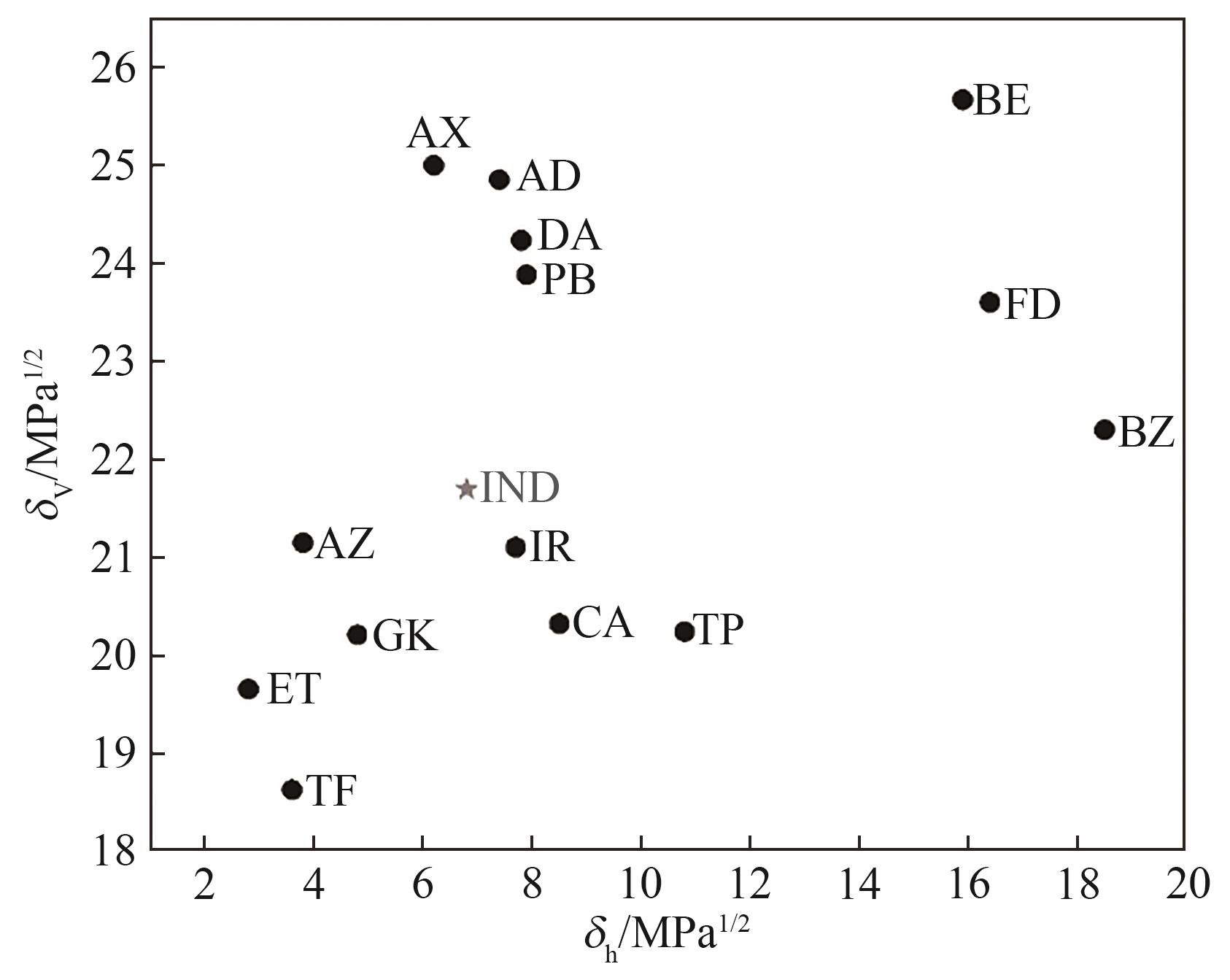
Fig.3 Bagley diagram of IND and drug candidates
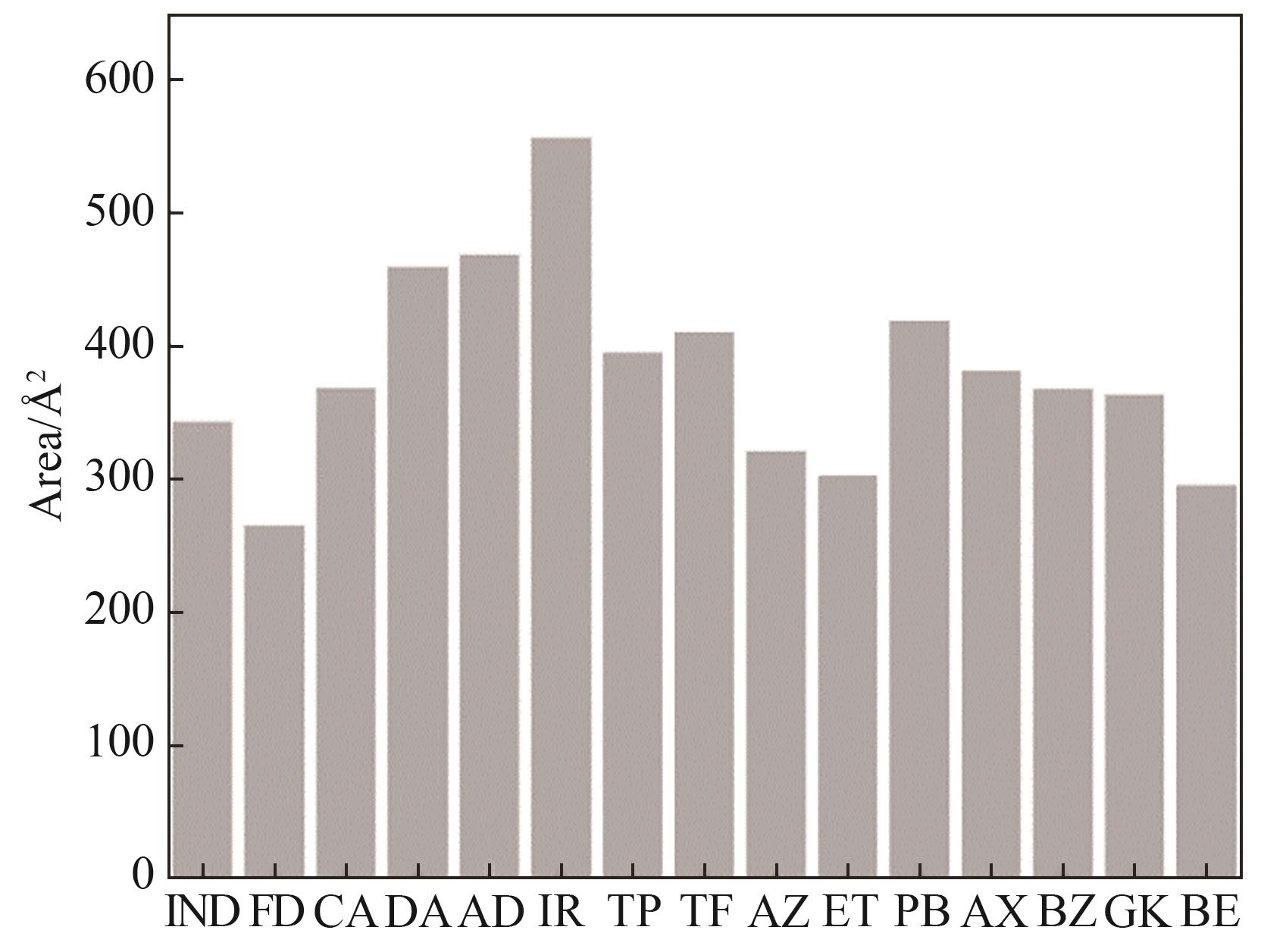
Fig.4 Charge surface area of drug molecules
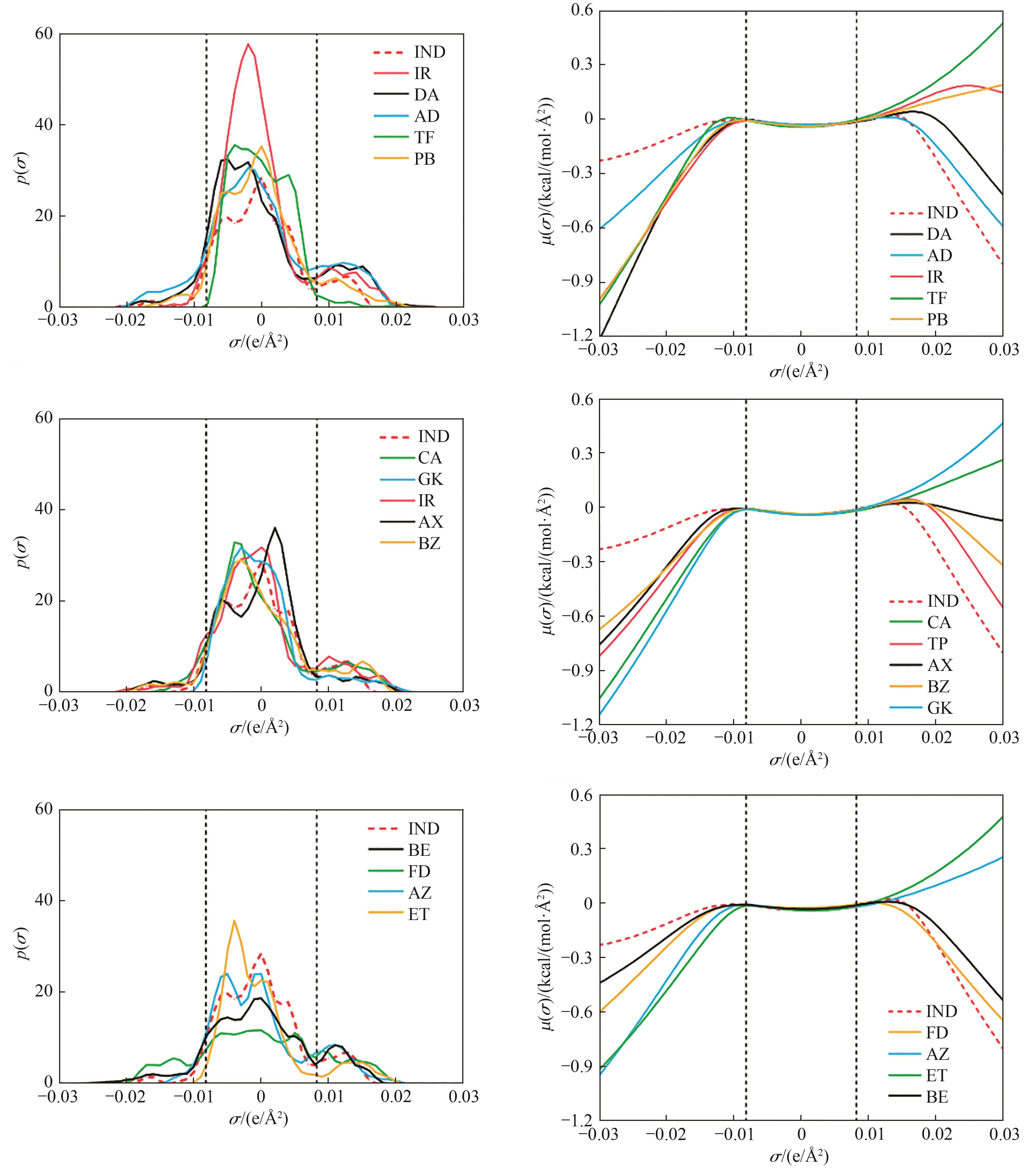
Fig.5 σ-profiles (left) and σ- potentials (right) of drug molecules
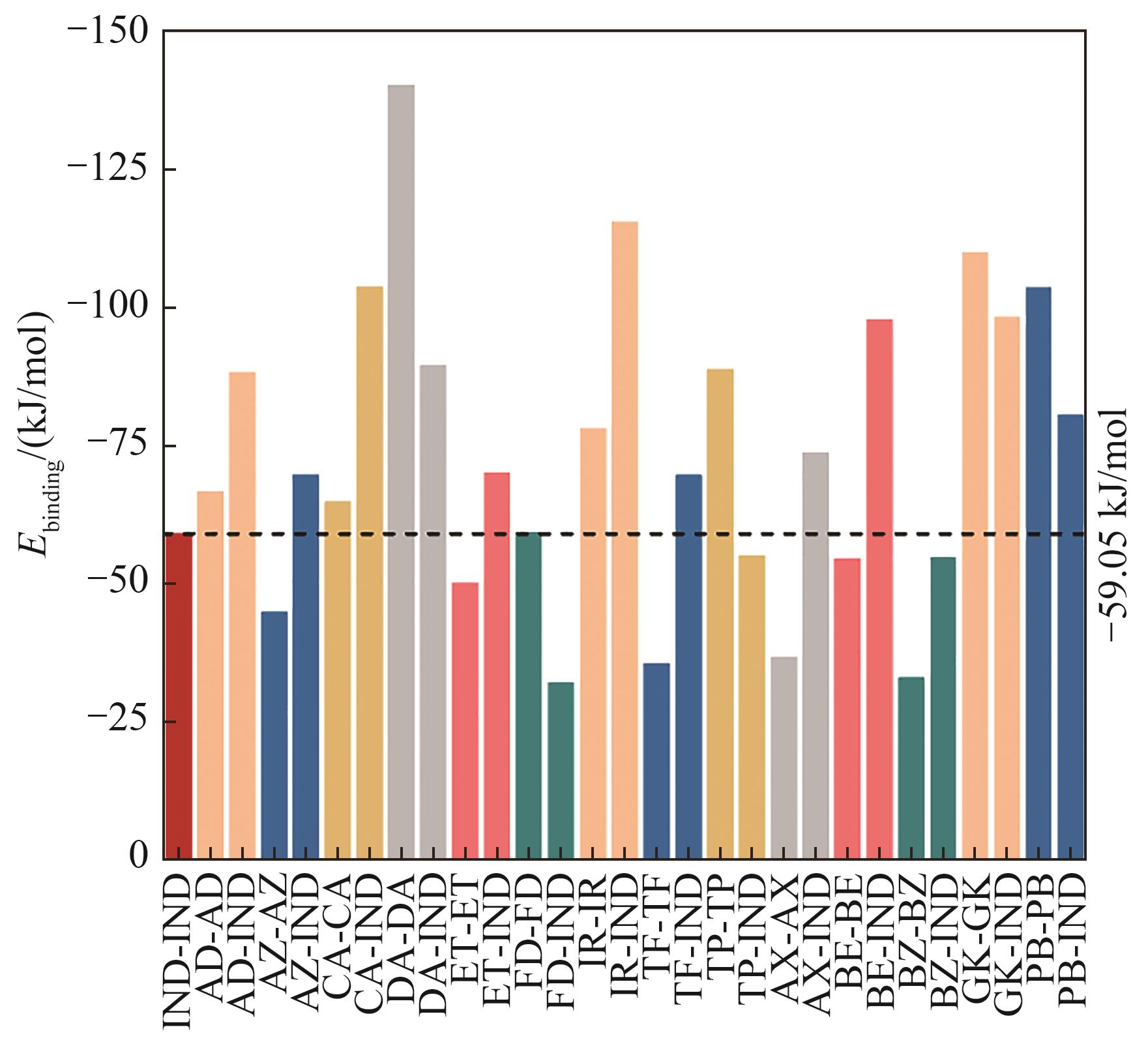
Fig.6 Binding energy of drug molecule self-aggregation and self-assembly with indomethacin
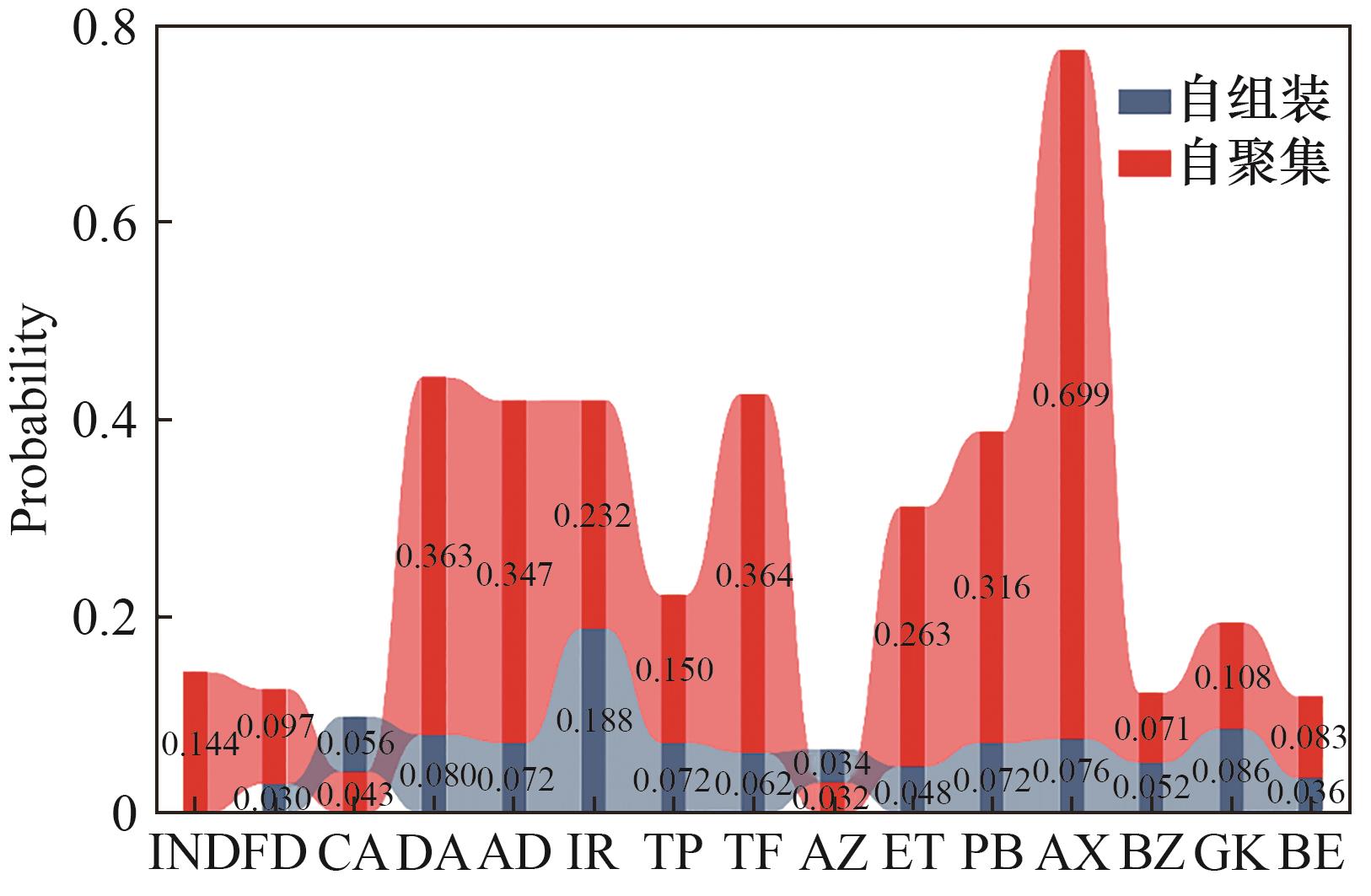
Fig.7 Probability of self-aggregation and self-assembly with indomethacin for the candidate drugs predicted by the ML model
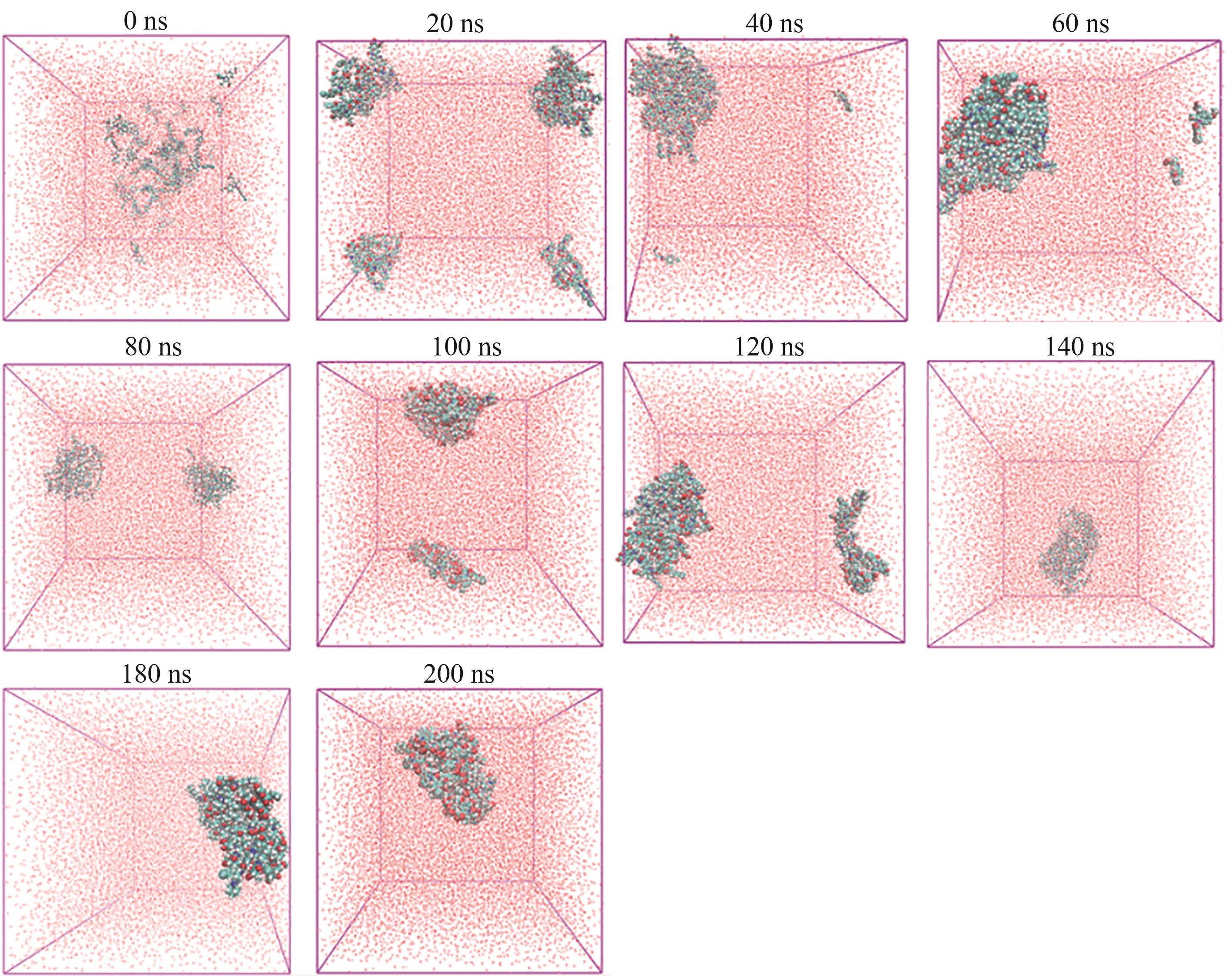
Fig.8 Molecular dynamics simulation snapshot of IR-IND
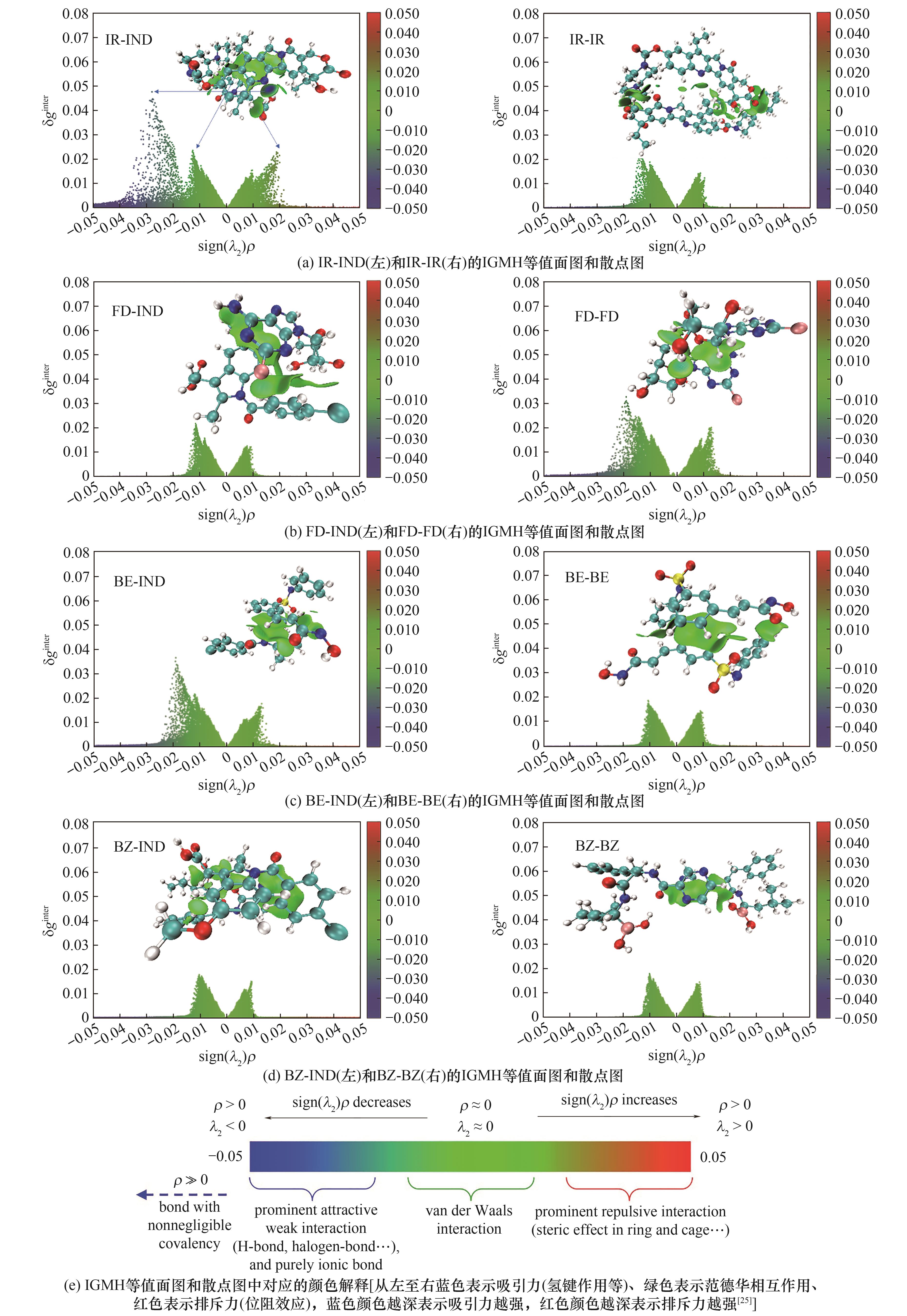
Fig.9 Schematic diagram of the results of intermolecular interactions between drugs analyzed by IGMH
| 1 | Sung H, Ferlay J, Siegel R L, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA: A Cancer Journal for Clinicians, 2021, 71(3): 209-249. |
| 2 | Xiao Y T, Liu J, Guo M Y, et al. Synergistic combination chemotherapy using carrier-free celastrol and doxorubicin nanocrystals for overcoming drug resistance[J]. Nanoscale, 2018, 10(26): 12639-12649. |
| 3 | Salvador-Morales C, Grodzinski P. Nanotechnology tools enabling biological discovery[J]. ACS Nano, 2022, 16(4): 5062-5084. |
| 4 | Liu K F, Liu Y X, Li C X, et al. Self-assembled pH and redox dual responsive carboxymethylcellulose-based polymeric nanoparticles for efficient anticancer drug codelivery[J]. ACS Biomaterials Science & Engineering, 2018, 4(12): 4200-4207. |
| 5 | Kianfar E. Protein nanoparticles in drug delivery: animal protein, plant proteins and protein cages, albumin nanoparticles[J]. Journal of Nanobiotechnology, 2021, 19(1): 159. |
| 6 | Liu Y B, Castro Bravo K M, Liu J W. Targeted liposomal drug delivery: a nanoscience and biophysical perspective[J]. Nanoscale Horizons, 2021, 6(2): 78-94. |
| 7 | Kheraldine H, Rachid O, Habib A M, et al. Emerging innate biological properties of nano-drug delivery systems: a focus on PAMAM dendrimers and their clinical potential[J]. Advanced Drug Delivery Reviews, 2021, 178: 113908. |
| 8 | Wang X W, Zhong X Y, Li J X, et al. Inorganic nanomaterials with rapid clearance for biomedical applications[J]. Chemical Society Reviews, 2021, 50(15): 8669-8742. |
| 9 | Kuang Y T, Li Z K, Chen H, et al. Advances in self-assembled nanotechnology in tumor therapy[J]. Colloids and Surfaces B: Biointerfaces, 2024, 237: 113838. |
| 10 | Zhang X B, Li N, Zhang S W, et al. Emerging carrier-free nanosystems based on molecular self-assembly of pure drugs for cancer therapy[J]. Medicinal Research Reviews, 2020, 40(5): 1754-1775. |
| 11 | 刘雨婷, 王悦全, 张申武, 等. 小分子自组装纳米递药系统研究进展[J]. 药学学报, 2023, 58(3): 516-529. |
| Liu Y T, Wang Y Q, Zhang S W, et al. Advance on small molecule self-assembled nano-drug delivery system[J]. Acta Pharmaceutica Sinica, 2023, 58(3): 516-529. | |
| 12 | Xu X L, Liu A, Liu S Q, et al. Application of molecular dynamics simulation in self-assembled cancer nanomedicine[J]. Biomaterials Research, 2023, 27(1): 39. |
| 13 | Fang F, Chen X Y. Carrier-free nanodrugs: from bench to bedside[J]. ACS Nano, 2024, 18(35): 23827-23841. |
| 14 | Zhong T, Hao Y L, Yao X, et al. Effect of XlogP and Hansen solubility parameters on small molecule modified paclitaxel anticancer drug conjugates self-assembled into nanoparticles[J]. Bioconjugate Chemistry, 2018, 29(2): 437-444. |
| 15 | Reker D, Rybakova Y, Kirtane A R, et al. Computationally guided high-throughput design of self-assembling drug nanoparticles[J]. Nature Nanotechnology, 2021, 16(6): 725-733. |
| 16 | Shamay Y, Shah J, Işık M, et al. Quantitative self-assembly prediction yields targeted nanomedicines[J]. Nature Materials, 2018, 17(4): 361-368. |
| 17 | Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 16 Rev. A.03 [CP]. Wallingford, CT, 2016. |
| 18 | Zhao Y, Truhlar D G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals[J]. Theoretical Chemistry Accounts, 2008, 120(1): 215-241. |
| 19 | Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu[J]. Journal of Chemical Physics, 2010, 132(15): 154104. |
| 20 | Klamt A, Schüürmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient[J]. Journal of the Chemical Society, Perkin Transactions 2, 1993(5): 799-805. |
| 21 | Xantheas S S. On the importance of the fragment relaxation energy terms in the estimation of the basis set superposition error correction to the intermolecular interaction energy[J]. The Journal of Chemical Physics, 1996, 104(21): 8821-8824. |
| 22 | Lu T, Chen F W. Multiwfn: a multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592. |
| 23 | Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics[J]. Journal of Molecular Graphics, 1996, 14(1): 33-38. |
| 24 | Lu T, Chen Q. Visualization analysis of weak interactions in chemical systems [M]//YáñEZ M, Boyd R J. Comprehensive Computational Chemistry. Oxford: Elsevier, 2024: 240-264. |
| 25 | Lu T, Chen Q X. Independent gradient model based on hirshfeld partition: a new method for visual study of interactions in chemical systems[J]. Journal of Computational Chemistry, 2022, 43(8): 539-555. |
| 26 | Abraham M J, Murtola T, Schulz R, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers[J]. SoftwareX, 2015, 1: 19-25. |
| 27 | Wang J M, Wolf R M, Caldwell J W, et al. Development and testing of a general amber force field[J]. Journal of Computational Chemistry, 2004, 25(9): 1157-1174. |
| 28 | Martínez L, Andrade R, Birgin E G, et al. PACKMOL: a package for building initial configurations for molecular dynamics simulations[J]. Journal of Computational Chemistry, 2009, 30(13): 2157-2164. |
| 29 | Berendsen H J C, Postma J P M, van Gunsteren W F, et al. Molecular dynamics with coupling to an external bath[J]. The Journal of Chemical Physics, 1984, 81(8): 3684-3690. |
| 30 | Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method[J]. Journal of Applied Physics, 1981, 52(12): 7182-7190. |
| 31 | Parrinello M, Rahman A. Strain fluctuations and elastic constants[J]. The Journal of Chemical Physics, 1982, 76(5): 2662-2666. |
| 32 | Hockney R W. The potential calculation and some applications[J]. Methods in Computational Physics, 1970, 9: 136-211. |
| 33 | Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems[J]. The Journal of Chemical Physics, 1993, 98(12): 10089-10092. |
| 34 | Essmann U, Perera L, Berkowitz M L, et al. A smooth particle mesh Ewald method[J]. The Journal of Chemical Physics, 1995, 103(19): 8577-8593. |
| 35 | Salem A, Nagy S, Pál S, et al. Reliability of the Hansen solubility parameters as co-crystal formation prediction tool[J]. International Journal of Pharmaceutics, 2019, 558: 319-327. |
| 36 | Shete A, Murthy S, Korpale S, et al. Cocrystals of itraconazole with amino acids: screening, synthesis, solid state characterization, in vitro drug release and antifungal activity[J]. Journal of Drug Delivery Science and Technology, 2015, 28: 46-55. |
| 37 | Mohammad M A, Alhalaweh A, Velaga S P. Hansen solubility parameter as a tool to predict cocrystal formation[J]. International Journal of Pharmaceutics, 2011, 407(1/2): 63-71. |
| 38 | Bagley E, Nelson T, Scigliano J. Three-dimensional solubility parameters and their relationship to internal pressure measurements in polar and hydrogen bonding solvents[J]. Journal of Paint Technology, 1971, 43(555): 35-42. |
| 39 | Breitkreutz J. Prediction of intestinal drug absorption properties by three-dimensional solubility parameters[J]. Pharmaceutical Research, 1998, 15(9): 1370-1375. |
| 40 | Klamt A. Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena[J]. The Journal of Physical Chemistry, 1995, 99(7): 2224-2235. |
| 41 | Loschen C, Klamt A. Solubility prediction, solvate and cocrystal screening as tools for rational crystal engineering[J]. Journal of Pharmacy and Pharmacology, 2015, 67(6): 803-811. |
| 42 | Klamt A, Eckert F, Hornig M, et al. Prediction of aqueous solubility of drugs and pesticides with COSMO-RS[J]. Journal of Computational Chemistry, 2002, 23(2): 275-281. |
| 43 | Eckert F, Klamt A. Fast solvent screening via quantum chemistry: COSMO-RS approach[J]. AIChE Journal, 2002, 48(2): 369-385. |
| 44 | 米泽豪, 花儿. 基于DFT和COSMO-RS理论研究多元胺型离子液体吸收SO2气体[J]. 化工学报, 2023, 74(9): 3681-3696. |
| Mi Z H, Hua E. DFT and COSMO-RS theoretical analysis of SO2 absorption by polyamines type ionic liquids[J]. CIESC Journal, 2023, 74(9): 3681-3696. | |
| 45 | Cao Z X, Wu X J, Wei X H. Ionic liquid screening for desulfurization of coke oven gas based on COSMO-SAC model and process simulation[J]. Chemical Engineering Research and Design, 2021, 176: 146-161. |
| 46 | Mohan M, Keasling J D, Simmons B A, et al. In silico COSMO-RS predictive screening of ionic liquids for the dissolution of plastic[J]. Green Chemistry, 2022, 24(10): 4140-4152. |
| [1] | Xinyue WANG, Xiaohu XU, Haiyang ZHANG, Chunhua YIN. Study on encapsulation and properties vitamin A acetate/cyclodextrin [J]. CIESC Journal, 2024, 75(S1): 321-328. |
| [2] | Zheming WU, Biyun ZHANG, Renchao ZHENG. Engineering of nitrilase enantioselectivity for efficient synthesis of brivaracetam [J]. CIESC Journal, 2024, 75(7): 2633-2643. |
| [3] | Hansong QIN, Guoliang LI, Hao YAN, Xiang FENG, Yibin LIU, Xiaobo CHEN, Chaohe YANG. Theoretical study on the adsorption and diffusion behavior of methyl oleate catalytic cracking in hierarchical ZSM-5 zeolite [J]. CIESC Journal, 2024, 75(5): 1870-1881. |
| [4] | Yiru WEN, Jia FU, Dahuan LIU. Advances in machine learning-based materials research for MOFs: energy gas adsorption separation [J]. CIESC Journal, 2024, 75(4): 1370-1381. |
| [5] | Kang ZHOU, Jianxin WANG, Hai YU, Chaoliang WEI, Fengqi FAN, Xinhao CHE, Lei ZHANG. Foam rupture properties of mineral base oils based on molecular dynamics simulation [J]. CIESC Journal, 2024, 75(4): 1668-1678. |
| [6] | Dongfei LIU, Fan ZHANG, Zheng LIU, Diannan LU. A review of machine learning potentials and their applications to molecular simulation [J]. CIESC Journal, 2024, 75(4): 1241-1255. |
| [7] | Zheng ZHANG, Wuqiong WANG, Yajing ZHANG, Kangjun WANG, Yuanhui JI. Research progress in theoretical calculation of pharmaceutical formulation design [J]. CIESC Journal, 2024, 75(4): 1429-1438. |
| [8] | Yujiao ZENG, Xin XIAO, Gang YANG, Yibo ZHANG, Guangming ZHENG, Fang LI, Fengling WANG. Surrogate modeling and optimization of wet phosphoric acid production process based on mechanism and data hybrid driven [J]. CIESC Journal, 2024, 75(3): 936-944. |
| [9] | Yuxiang CHEN, Chuanlei LIU, Zijun GONG, Qiyue ZHAO, Guanchu GUO, Hao JIANG, Hui SUN, Benxian SHEN. Machine learning-assisted solvent molecule design for efficient absorption of ethanethiol [J]. CIESC Journal, 2024, 75(3): 914-923. |
| [10] | Fan WU, Xudong PENG, Jinbo JIANG, Xiangkai MENG, Yangyang LIANG. Study on adaptability of molecular dynamics in predicting density and viscosity of natural gas [J]. CIESC Journal, 2024, 75(2): 450-462. |
| [11] | Jian RUAN, Shuang LI, Zhenghui WEN. Application of automation and artificial intelligence in flow chemistry [J]. CIESC Journal, 2024, 75(11): 4120-4140. |
| [12] | Maoxian WANG, Qidian SUN, Zhe FU, Fang HUA, Ye JI, Yi CHENG. Understanding pyrolysis process of polyethylene by combined method of molecular-level kinetic model with machine learning [J]. CIESC Journal, 2024, 75(11): 4320-4332. |
| [13] | Han TANG, Jin CAI, Haihang QIN, Guangjin CHEN, Changyu SUN. Predictive model on gas solubility in water-rich phase coexisted with gas hydrates [J]. CIESC Journal, 2024, 75(11): 4348-4358. |
| [14] | Gen LIU, Zhongshun SUN, Bo ZHANG, Rongjiang ZHANG, Zhiqiang WU, Bolun YANG. Establishment of machine learning-driven biomass pyrolysis model and optimization of volatiles chemical looping reforming hydrogen production process [J]. CIESC Journal, 2024, 75(11): 4333-4347. |
| [15] | Min FU, Zijian CHEN, Shuai TANG, Xiliang QIAN, Zengxi WEI, Yun ZOU, Zhangfa TONG. Molecular simulation of small molecule adsorption and diffusion behavior in PVA hybrid membranes [J]. CIESC Journal, 2024, 75(11): 4152-4161. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

