化工学报 ›› 2020, Vol. 71 ›› Issue (1): 192-199.DOI: 10.11949/0438-1157.20191229
吴杰( ),李嘉辉,于燕梅,于养信()
),李嘉辉,于燕梅,于养信()
收稿日期:2019-09-23
修回日期:2019-10-24
出版日期:2020-01-05
发布日期:2020-01-05
通讯作者:
于养信
作者简介:吴杰(1995—),男,博士研究生,基金资助:
Jie WU(),Jiahui LI,Yanmei YU,Yangxin YU()
Received:2019-09-23
Revised:2019-10-24
Online:2020-01-05
Published:2020-01-05
Contact:
Yangxin YU
摘要:
第Ⅲ族元素磷化物是重要的半导体材料,其在有限温度下热力学性质是该类材料设计和应用的基础。在密度泛函理论以及声子模型的基础上,系统研究了第Ⅲ族元素磷化物BP、AlP、GaP和InP的结构性质及其在0~1000 K温度范围内的热膨胀系数和热力学函数。计算结果表明,四种磷化物的晶胞参数计算值与已有的实验值符合得很好。通过非简谐近似计算,发现这四种磷化物的焓、熵、热膨胀系数这些热力学性质会随着温度的升高基本呈现单调递增变化,其热容则在增加到一定程度后趋于Dulong-Petit极限。此外,还将热力学函数计算值与可提供的实验值进行了比较,发现两者一致性较好。
中图分类号:
吴杰, 李嘉辉, 于燕梅, 于养信. 第Ⅲ族元素磷化物热力学性质理论研究[J]. 化工学报, 2020, 71(1): 192-199.
Jie WU, Jiahui LI, Yanmei YU, Yangxin YU. Theoretical investigation on thermodynamic properties of group Ⅲ phosphides[J]. CIESC Journal, 2020, 71(1): 192-199.
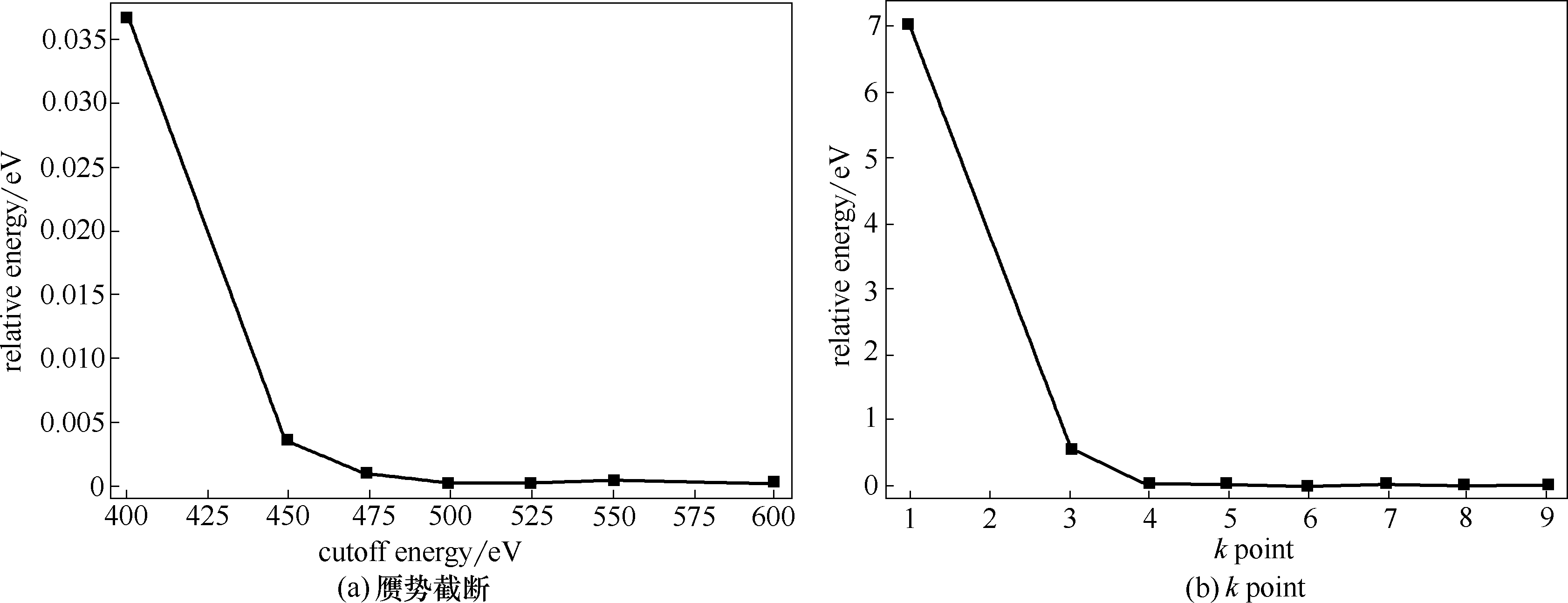
图1 收敛性测试
Fig.1 Convergence test
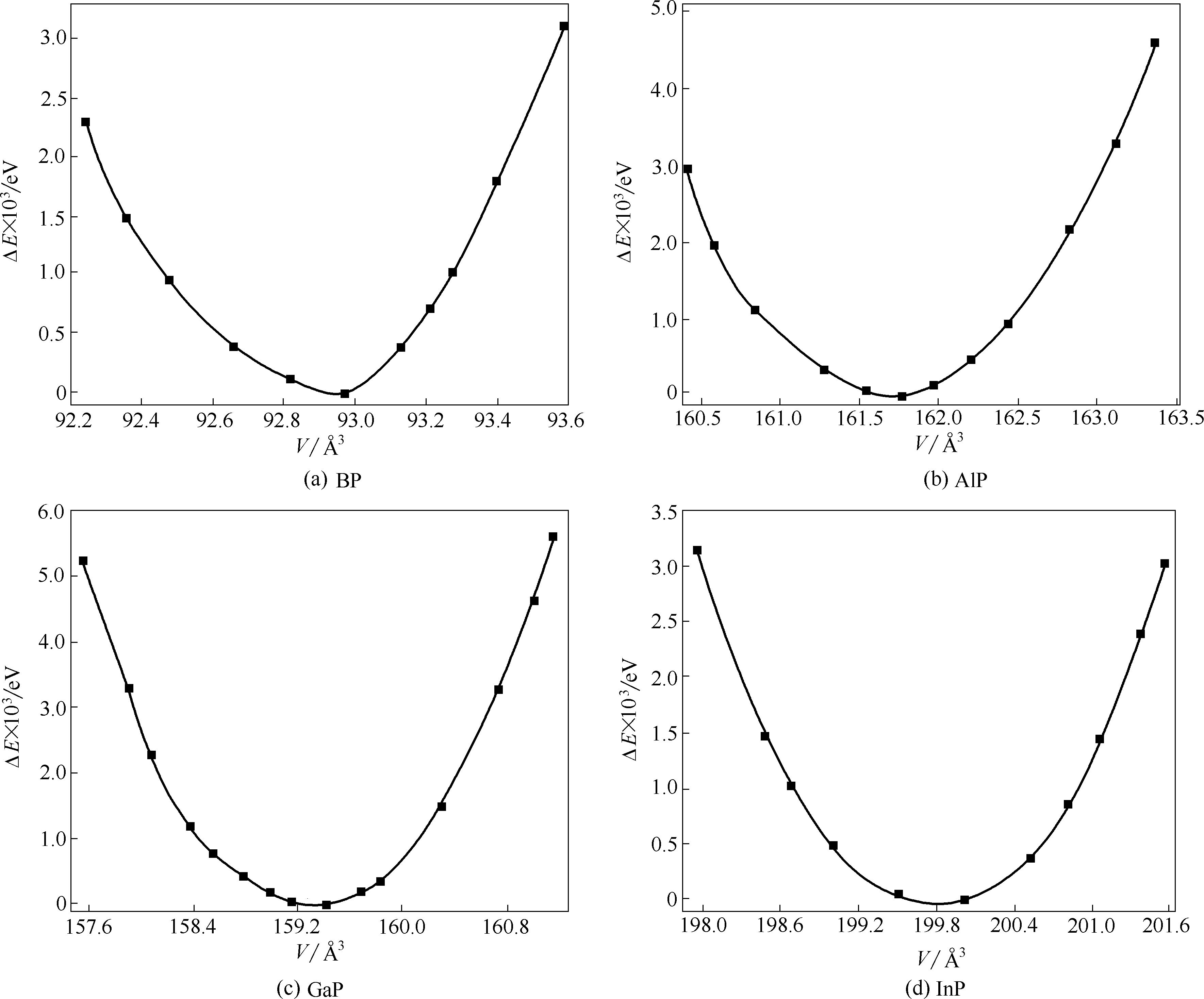
图2 第Ⅲ族元素磷化物的相对能量计算值(0 K)随体积变化曲线
Fig.2 Calculated relative energy (0 K) for group Ⅲ phosphides as a function of volume
| 磷化物 | M-P 键长/ ? | 晶胞边长a/? | 体积模量B/GPa | ||||
|---|---|---|---|---|---|---|---|
| 本文 | 文献 | ||||||
| 0 K | 300 K | 实验(300 K) | 其他计算(0 K) | 本文 | 实验 | ||
| BP AlP GaP InP | 1.96 2.36 2.34 2.53 | 4.530 5.449 5.422 5.848 | 4.531 5.450 5.427 5.850 | 4.54[ 5.45[ 5.45[ 5.87[ | 4.55[ 5.53[ 5.52[ 5.95[ | 161.3 86.7 84.8 64.8 | 173[ 86[ 91[ 72[ |
表1 闪锌矿结构金属磷化物的键长、晶胞边长和体积模量
Table 1 Bond lengths, lattice constants a and bulk modulus for zinc-blende metal phosphides
| 磷化物 | M-P 键长/ ? | 晶胞边长a/? | 体积模量B/GPa | ||||
|---|---|---|---|---|---|---|---|
| 本文 | 文献 | ||||||
| 0 K | 300 K | 实验(300 K) | 其他计算(0 K) | 本文 | 实验 | ||
| BP AlP GaP InP | 1.96 2.36 2.34 2.53 | 4.530 5.449 5.422 5.848 | 4.531 5.450 5.427 5.850 | 4.54[ 5.45[ 5.45[ 5.87[ | 4.55[ 5.53[ 5.52[ 5.95[ | 161.3 86.7 84.8 64.8 | 173[ 86[ 91[ 72[ |
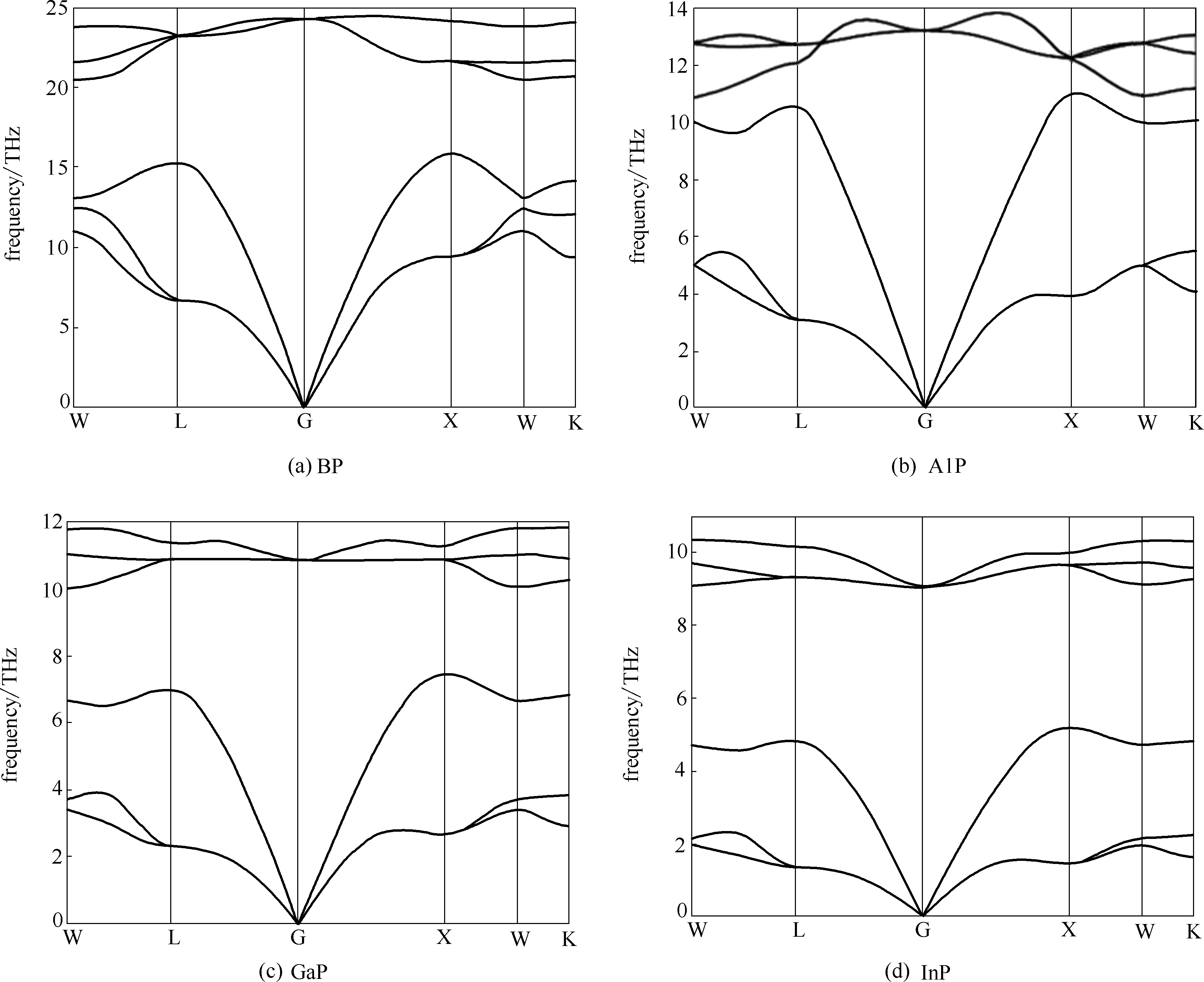
图3 第Ⅲ族元素磷化物在0 K时的声子色散曲线
Fig.3 Calculated phonon dispersion curves for group Ⅲ phosphides at 0 K
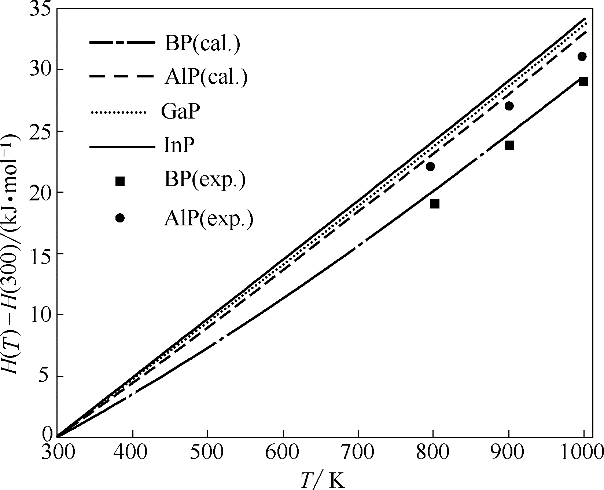
图4 BP、AlP、GaP和InP的焓随温度的变化(H(300)表示300 K时物质的焓)
Fig.4 Calculated enthalpy as a function of temperature for BP, AlP, GaP and InP (H(300) is enthalpy at 300 K)
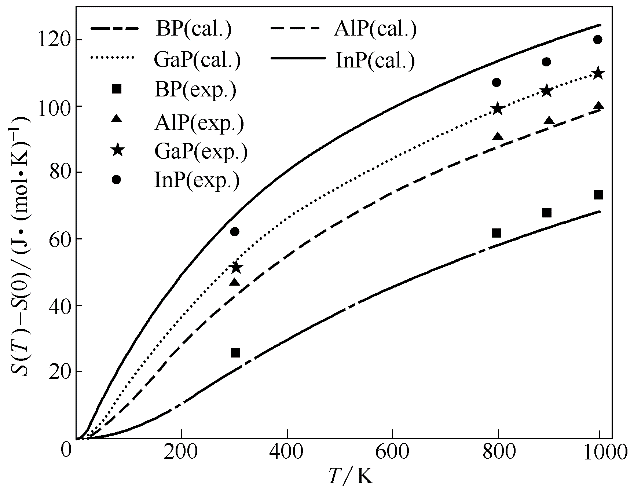
图5 BP、AlP、GaP和InP的熵随温度的变化(S(0)表示0 K时物质的熵)
Fig.5 Calculated entropy as a function of temperature for BP, AlP, GaP and InP (S(0) is entropy at 0 K)
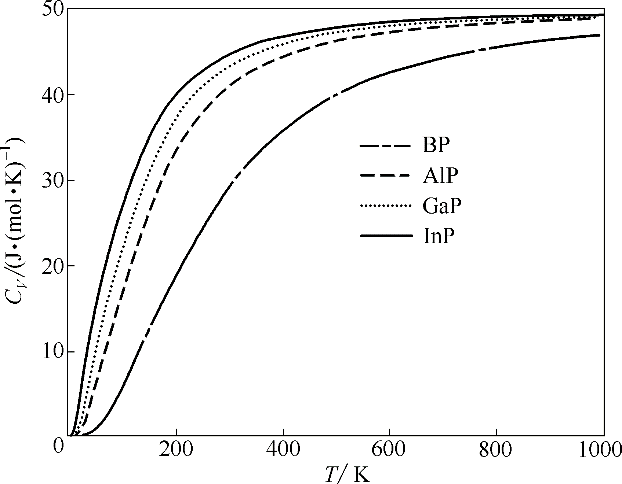
图6 BP、AlP、GaP和InP的计算热容随温度的变化
Fig.6 Calculated heat capacity as a function of temperature for BP, AlP, GaP and InP
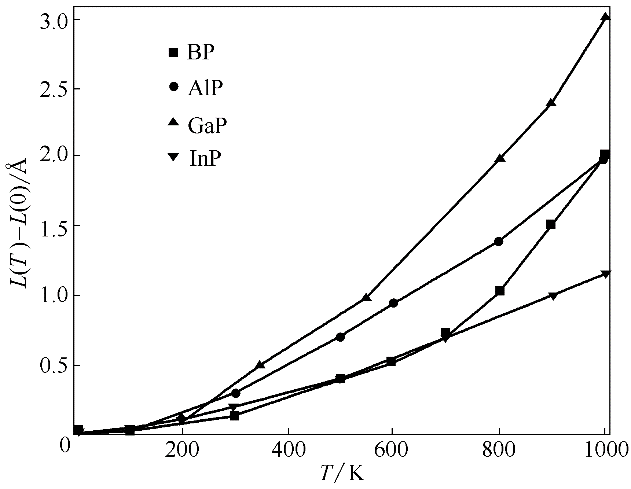
图7 BP、AlP、GaP和InP的平衡晶胞边长随温度的变化(L(0)表示0 K时晶胞平衡边长)
Fig.7 Calculated equilibrium lattice parameter as a function of temperature for BP, AlP, GaP and InP (L(0) is equilibrium lattice parameter at 0 K)
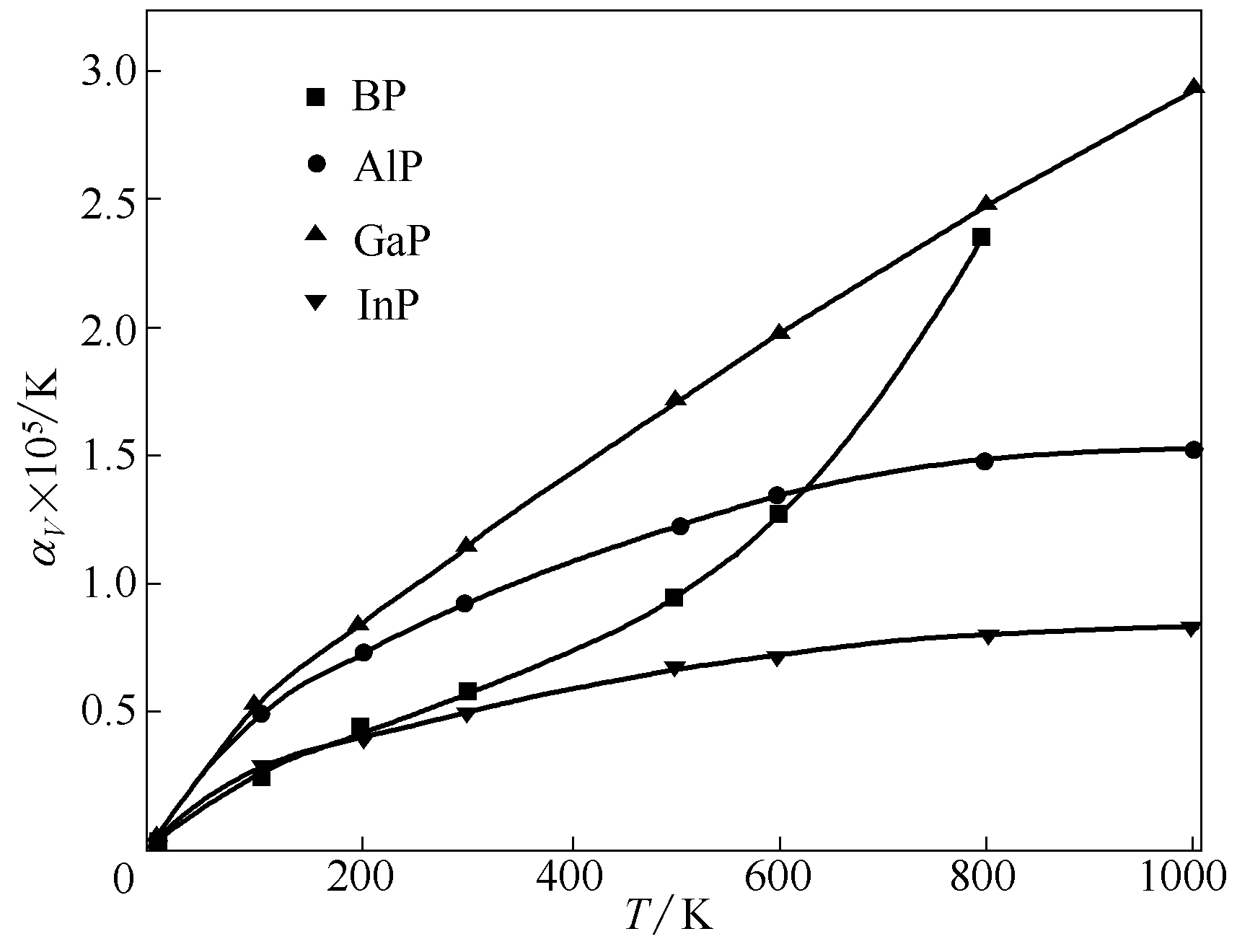
图8 BP、AlP、GaP和InP的体积热膨胀系数随温度的变化
Fig.8 Calculated volume thermal-expansion coefficient as a function of temperature for BP, AlP, GaP and InP
| 1 | Vurgaftman I , Meyer J R , Ram-Mohan L R . Band parameters for Ⅲ-V compound semiconductors and their alloys[J]. Journal of Applied Physics, 2001, 89(11): 5815-5875. |
| 2 | Yu L H , Yao K L , Liu Z L . Electronic band structures of filled tetrahedral semiconductor LiMgP and zinc-blende AlP[J]. Solid State Communications, 2005, 135(1/2): 124-128. |
| 3 | Zaoui A , Kacimi S , Yakoubi A , et al . Optical properties of BP, BAs and BSb compounds under hydrostatic pressure[J]. Physica B-Condensed Matter, 2005, 367(1/2/3/4): 195-204. |
| 4 | Touat D , Ferhat M , Zaoui A . Dynamical behaviour in the boron Ⅲ-Ⅴ group: a first-principles study[J]. Journal of Physics-Condensed Matter, 2006, 18(15): 3647-3654. |
| 5 | Lu X F , Gao X , Li C X , et al . Electronic structure and optical properties of doped gallium phosphide: a first-principles simulation[J]. Physics Letters A, 2017, 381(35): 2986-2992. |
| 6 | Tong C J , Zhang H , Zhang Y N , et al . New manifold two-dimensional single-layer structures of zinc-blende compounds[J]. Journal of Materials Chemistry A, 2014, 2(42): 17971-17978. |
| 7 | Fornari R . Single Crystals of Electronic Materials: Growth and Properties[M]. Cambridge: Woodhead Publ. Ltd., 2019: 241-268. |
| 8 | Crisp R W , Kirkwood N , Grimaldi G , et al . Highly photoconductive InP quantum dots films and solar cells[J]. ACS Applied Energy Materials, 2018, 1(11): 6569-6576. |
| 9 | Li S , Taddei K M , Wang X Q , et al . Thermal expansion coefficients of high thermal conducting BAs and BP materials[J]. Applied Physics Letters, 2019, 115(1): 011901. |
| 10 | Abrishamifar S M , Heidari N , Razavi R , et al . The Cl functionalized aluminum nitride (AlN) and aluminum phosphide (AlP) nanocone sheets as hydrogen selenide (H2Se) sensor: a density functional investigation[J]. Acta Chimica Slovenica, 2018, 65(1): 208-212. |
| 11 | Chen Z H , Shao Z H , Siddiqui M K , et al . Potential of carbon, silicon, boron nitride and aluminum phosphide nanocages as anodes of lithium, sodium and potassium ion batteries: a DFT study[J]. Russian Journal of Physical Chemistry B, 2019, 13(1): 156-164. |
| 12 | Halmann M . Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells[J]. Nature, 1978, 275(5676): 115-116. |
| 13 | Jiao Z Y , Ma S H , Guo Y L . Simulation of optical function for phosphide crystals following the DFT band structure calculations[J]. Computational and Theoretical Chemistry, 2011, 970(1/2/3): 79-84. |
| 14 | Britto R J , Young J L , Yang Y , et al . Interfacial engineering of gallium indium phosphide photoelectrodes for hydrogen evolution with precious metal and non-precious metal based catalysts[J]. Journal of Materials Chemistry A, 2019, 7(28): 16821-16832. |
| 15 | Hestroffer K , Sperlich D , Dadgostar S , et al . Transport properties of doped AlP for the development of conductive AlP/GaP distributed Bragg reflectors and their integration into light-emitting diodes[J]. Applied Physics Letters, 2018, 112(19): 191207. |
| 16 | Zhao S , Butera S , Lioliou G , et al . AlInP photodiode X-ray detectors[J]. Journal of Physics D-Applied Physics, 2019, 52(22): 225101. |
| 17 | Mujica A , Rubio A , Munoz A , et al . High-pressure phases of group-IV, Ⅲ-V, and II-VI compounds[J]. Reviews of Modern Physics, 2003, 75(3): 863-912. |
| 18 | Saib S , Bouarissa N , Rodriguez-Hernandez P , et al . Elastic modulus and thermal properties of InN in the rocksalt phase[J]. Computational Materials Science, 2014, 81: 374-377. |
| 19 | Bioud N , Kassali K , Bouarissa N . Thermodynamic properties of compressed CuX (X = Cl, Br) compounds: ab initio study[J]. Journal of Electronic Materials, 2017, 46(4): 2521-2528. |
| 20 | Daoud S , Bouarissa N . Structural and thermodynamic properties of cubic sphalerite aluminum nitride under hydrostatic compression[J]. Computational Condensed Matter, 2019, 19: e00359. |
| 21 | Daoud S , Bouarissa N , Bioud N , et al . High-temperature and high-pressure thermophysical properties of AlP semiconducting material: a systematic ab initio study[J]. Chemical Physics, 2019, 525: UNSP 110399. |
| 22 | Settouf A , Rached H , Benkhettou N , et al . DFT calculations of structural, optoelectronic and thermodynamic properties of B x Al1 -x P alloys[J]. Computational Condensed Matter, 2019, 19: e00377. |
| 23 | Segall M D , Lindan P J D , Probert M J , et al . First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics-Condensed Matter, 2002, 14(11): 2717-2744. |
| 24 | Lejaeghere K , Bihlmayer G , Björkman T , et al . Reproducibility in density functional theory calculations of solids[J]. Science, 2016, 351(6280): aad3000. |
| 25 | Perdew J P , Burke K , Ernzerhof M . Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868. |
| 26 | Grimme S . Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J]. Journal of Computational Chemistry, 2006, 27(15): 1787-1799. |
| 27 | Baroni S , de Gironcoli S , Dal Corso A , et al . Phonons and related crystal properties from density-functional perturbation theory[J]. Reviews of Modern Physics, 2001, 73(2): 515-562. |
| 28 | Wyckoff R W G . Crystal Structures[M]. 2nd ed. New York: John Wiley & Sons Inc., 1964: 110-112. |
| 29 | Xia H , Xia Q , Ruoff A L . BP at megabar pressures and its equation of state to 110 GPa[J]. Journal of Applied Physics, 1993, 74(3): 1660-1662. |
| 30 | Lambrecht W R L , Segall B . Electronic-structure and bonding at SiC/AlN and SiC/BP interfaces[J]. Physical Review B, 1991, 43(9): 7070-7085. |
| 31 | Kalvoda S , Paulus B , Fulde P , et al . Influence of electron correlations on ground-state properties of Ⅲ-V semiconductors[J]. Physical Review B, 1997, 55(7): 4027-4030. |
| 32 | Wang S Q , Ye H Q . A plane-wave pseudopotential study on Ⅲ-V zinc-blende and wurtzite semiconductors under pressure[J]. Journal of Physics-Condensed Matter, 2002, 14(41): 9579-9587. |
| 33 | Zafar M , Masood M K , Rizwan M , et al . Theoretical study of structural, electronic, optical and elastic properties of Al x Ga 1-x P[J]. Optik, 2019, 182: 1176-1185. |
| 34 | Ahmed R , Fazal E A , Hashemifar S J , et al . First-principles study of the structural and electronic properties of Ⅲ-phosphides[J]. Physica B-Condensed Matter, 2008, 403(10/11): 1876-1881. |
| 35 | Vancamp P E , Vandoren V E , Devreese J T . Pressure-dependence of the electronic-properties of cubic Ⅲ-V in compounds[J]. Physical Review B, 1990, 41(3): 1598-1602. |
| 36 | Schroten E , Goossens A , Schoonman J . Photo- and electroreflectance of cubic boron phosphide[J]. Journal of Applied Physics, 1998, 83(3): 1660-1663. |
| 37 | Madelung O . Semiconductors: Data Handbook[M]. Berlin: Springer, 2004. |
| 38 | Lu Y L , Jia D W , Gao F , et al . First-principle calculations of the thermal properties of SrTiO3 and SrO(SrTiO3) n (n=1,2)[J]. Solid State Communications, 2015, 201: 25-30. |
| 39 | Dumont H , Montell Y . Some aspects on thermodynamic properties, phase diagram and alloy formation in the ternary system BAs-GaAs(Ⅰ): Analysis of BAs thermodynamic properties[J]. Journal of Crystal Growth, 2006, 290(2): 410-418. |
| 40 | Hou B S , Liu K , Mao X C , et al . Theoretical calculations for elastic and thermodynamic properties of NbN2 under high pressure[J]. Acta Physica Polonica A, 2017, 132(4): 1363-1370. |
| 41 | Lebga N , Daoud S , Sun X W , et al . Mechanical and thermophysical properties of cubic rock-salt AlN under high pressure[J]. Journal of Electronic Materials, 2018, 47(7): 3430-3439. |
| 42 | Ö Çiftci Y , Çolakoğlu K , Deligöz E , et al . First-principles calculations on structure, elastic and thermodynamic properties of Al2X (X=Sc, Y) under pressure[J]. Journal of Materials Science & Technology, 2012, 28(2): 155-163. |
| 43 | Xing M , Li B , Yu Z , et al . Elastic anisotropic and thermodynamic properties of I-4m2-BCN[J]. Acta Physica Polonica A, 2016, 129(6): 1124-1130. |
| 44 | Bouhafs B , Aourag H , Certier M . Trends in band-gap pressure coefficients in boron compounds BP, BAs, and BSb[J]. Journal of Physics: Condensed Matter, 2000, 12(26): 5655-5668. |
| [1] | 王光, 单发顺, 钱禹丞, 焦建芳. 基于集成学习传递熵的化工过程微小故障检测方法[J]. 化工学报, 2023, 74(7): 2967-2978. |
| [2] | 姚晓宇, 沈俊, 李健, 李振兴, 康慧芳, 唐博, 董学强, 公茂琼. 流体气液临界参数测量方法研究进展[J]. 化工学报, 2023, 74(5): 1847-1861. |
| [3] | 雷博雯, 吴建华, 吴启航. R290低压比热泵高补气过热度循环研究[J]. 化工学报, 2023, 74(5): 1875-1883. |
| [4] | 陈科, 杜理, 曾英, 任思颖, 于旭东. 四元体系LiCl+MgCl2+CaCl2+H2O 323.2 K相平衡研究及计算[J]. 化工学报, 2023, 74(5): 1896-1903. |
| [5] | 毛元敬, 杨智, 莫松平, 郭浩, 陈颖, 罗向龙, 陈健勇, 梁颖宗. C6~C10烷醇的SAFT-VR Mie状态方程参数回归及其热物性研究[J]. 化工学报, 2023, 74(3): 1033-1041. |
| [6] | 程文婷, 李杰, 徐丽, 程芳琴, 刘国际. AlCl3·6H2O在FeCl3、CaCl2、KCl及KCl–FeCl3溶液中溶解度的实验及预测[J]. 化工学报, 2023, 74(2): 642-652. |
| [7] | 杨松涛, 李东洋, 牛玉清, 李鑫钢, 康绍辉, 李洪, 叶开凯, 周志全, 高鑫. 氟化物势能函数和热力学性质的分子模拟研究进展[J]. 化工学报, 2022, 73(9): 3828-3840. |
| [8] | 郭金玉, 王哲, 李元. 基于核熵独立成分分析的故障检测方法[J]. 化工学报, 2022, 73(8): 3647-3658. |
| [9] | 孙哲, 金华强, 李康, 顾江萍, 黄跃进, 沈希. 基于知识数据化表达的制冷空调系统故障诊断方法[J]. 化工学报, 2022, 73(7): 3131-3144. |
| [10] | 任嘉辉, 刘豫, 刘朝, 刘浪, 李莹. 基于分子指纹和拓扑指数的工质临界温度理论预测[J]. 化工学报, 2022, 73(4): 1493-1500. |
| [11] | 孙铭泽, 马宁, 李浩然, 姜海峰, 洪文鹏, 牛晓娟. 中低温超临界CO2及其混合工质布雷顿循环热力学分析[J]. 化工学报, 2022, 73(3): 1379-1388. |
| [12] | 许昊, 陈伟, 李邹路. 以[Li(TX-7)]SCN/H2O为工质对的第二类热泵特性研究[J]. 化工学报, 2022, 73(2): 577-586. |
| [13] | 王朋朋, 贾洋刚, 邵霞, 程婕, 冒爱琴, 檀杰, 方道来. K+掺杂尖晶石型(Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4高熵氧化物负极材料制备与储锂性能研究[J]. 化工学报, 2022, 73(12): 5625-5637. |
| [14] | 李怀旭, 孙晓岩, 陶少辉, 夏力, 项曙光. 基于分子热力学性质和密度峰聚类的脱硫汽油集总[J]. 化工学报, 2022, 73(12): 5449-5460. |
| [15] | 高腾飞, 李国选, 雷志刚. 从催化裂化柴油中分离联苯的溶剂筛选:实验和计算热力学[J]. 化工学报, 2022, 73(12): 5314-5323. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
 京公网安备 11010102001995号
京公网安备 11010102001995号

