化工学报 ›› 2022, Vol. 73 ›› Issue (9): 3828-3840.DOI: 10.11949/0438-1157.20220575
杨松涛1( ), 李东洋2(), 牛玉清4, 李鑫钢1, 康绍辉4, 李洪1, 叶开凯4, 周志全4, 高鑫1,3()
), 李东洋2(), 牛玉清4, 李鑫钢1, 康绍辉4, 李洪1, 叶开凯4, 周志全4, 高鑫1,3()
收稿日期:2022-04-24
修回日期:2022-07-01
出版日期:2022-09-05
发布日期:2022-10-09
通讯作者:
李东洋,高鑫
作者简介:杨松涛(1997—),男,硕士研究生,yangsongtao@tju.edu.cn
基金资助:
Songtao YANG1(), Dongyang LI2(), Yuqing NIU4, Xingang LI1, Shaohui KANG4, Hong LI1, Kaikai YE4, Zhiquan ZHOU4, Xin GAO1,3()
Received:2022-04-24
Revised:2022-07-01
Online:2022-09-05
Published:2022-10-09
Contact:
Dongyang LI, Xin GAO
摘要:
发展干法铀纯化工艺对我国核能产业发展至关重要,但一直受基础热力学数据缺失的限制。因分子模拟方法具有高效、环保、经济的预测特性,为解决以上挑战提供了机遇。本文归纳总结了分子模拟计算物质热力学性质的多种方法,系统地介绍了模拟计算中不同力场和势能函数的发展情况和优势/缺陷,指明了对氟化物体系的适用性。随后,综述了氟化物热力学性质的分子模拟进展,分析并评价了力场与势能函数选择对计算结果的影响,表明与温度相关的分子间势能函数(TDIP)在第二维里系数、黏度和气液共存性质等的模拟中展现的较高模拟效率和准确性。为进一步提升计算数据的准确性,尚有很多基础科学问题需要解决。因此,本文最后对分子模拟方法在氟化物热力学领域的发展进行了展望,以期为工艺设计提供实践基础。
中图分类号:
杨松涛, 李东洋, 牛玉清, 李鑫钢, 康绍辉, 李洪, 叶开凯, 周志全, 高鑫. 氟化物势能函数和热力学性质的分子模拟研究进展[J]. 化工学报, 2022, 73(9): 3828-3840.
Songtao YANG, Dongyang LI, Yuqing NIU, Xingang LI, Shaohui KANG, Hong LI, Kaikai YE, Zhiquan ZHOU, Xin GAO. Molecular simulation progress in studying thermodynamic properties and potential functions of fluorides[J]. CIESC Journal, 2022, 73(9): 3828-3840.

图1 氟化物的分子构型、物性及工业应用
Fig.1 Molecular configuration, physical properties and industrial applications of fluorides
| 分类 | 方法 | 特点 |
|---|---|---|
| 直接模拟法 | GEMC | 适合简单流体和较复杂流体 |
| CBMC | 适合复杂大分子或含有氢键等强相互作用的稠密流体 | |
| RGEMC | 适合模拟反应体系的相平衡 | |
| 间接模拟法 | NPT+TP | 模拟时间长,较为烦琐,应用较少 |
| GCMC | 适合非均相系统 | |
| GDI | 主要用于纯物质的计算,应用较多 | |
| HRW | 气液相波动剧烈的临界区的模拟 | |
| 新方法 | HRW和GEMC相结合 | 尚不成熟 |
| kMC | 适合模拟稀薄流体和缔合流体的相平衡 | |
| Bin-CMC | 气固相平衡 |
表1 Monte Carlo法的分类及特点
Table 1 Classification and characteristics of Monte Carlo method
| 分类 | 方法 | 特点 |
|---|---|---|
| 直接模拟法 | GEMC | 适合简单流体和较复杂流体 |
| CBMC | 适合复杂大分子或含有氢键等强相互作用的稠密流体 | |
| RGEMC | 适合模拟反应体系的相平衡 | |
| 间接模拟法 | NPT+TP | 模拟时间长,较为烦琐,应用较少 |
| GCMC | 适合非均相系统 | |
| GDI | 主要用于纯物质的计算,应用较多 | |
| HRW | 气液相波动剧烈的临界区的模拟 | |
| 新方法 | HRW和GEMC相结合 | 尚不成熟 |
| kMC | 适合模拟稀薄流体和缔合流体的相平衡 | |
| Bin-CMC | 气固相平衡 |
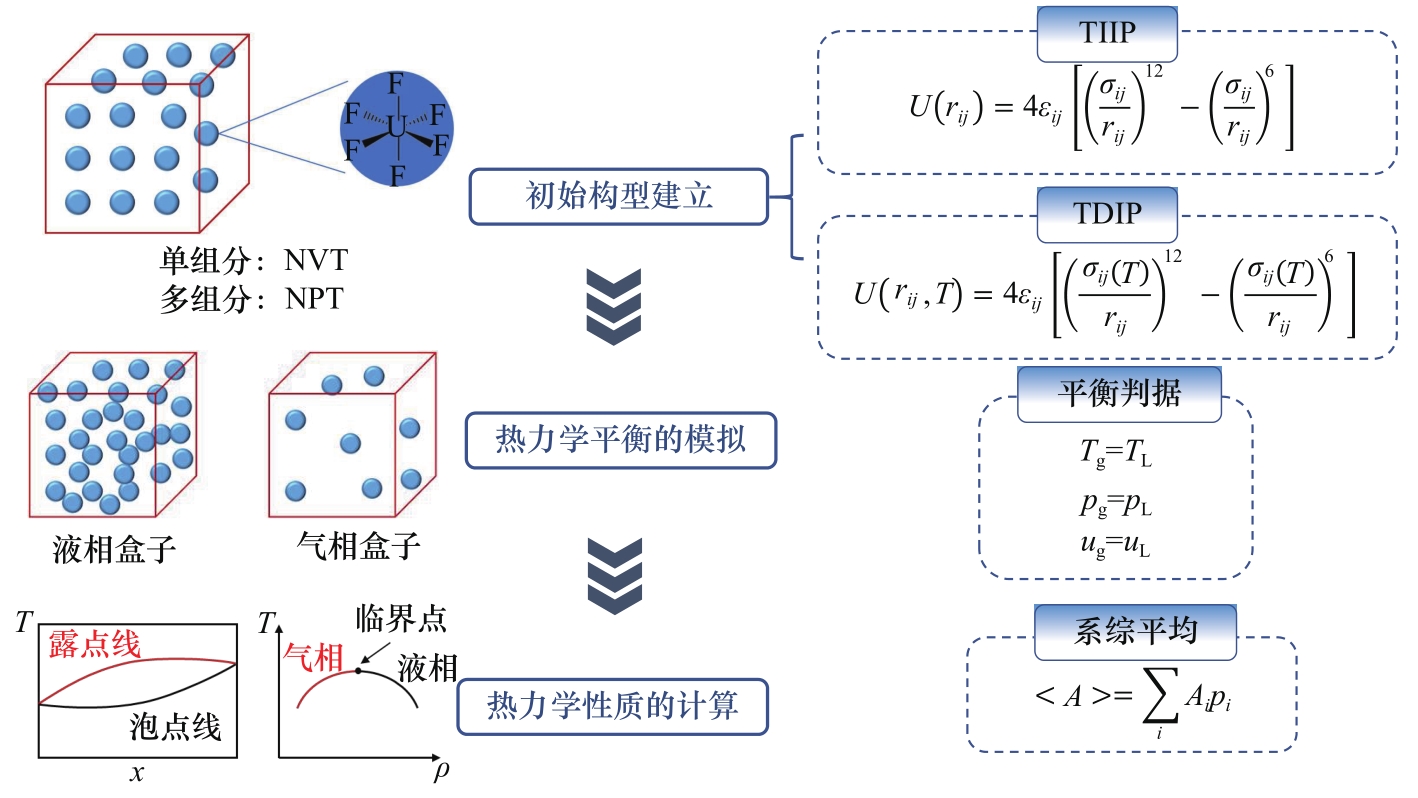
图2 氟化物相平衡热力学性质的GEMC方法
Fig.2 GEMC method for thermodynamic properties of fluoride phase equilibrium

图3 Lennard-Jones(n,6)势能曲线
Fig.3 Lennard-Jones (n, 6) potential curves
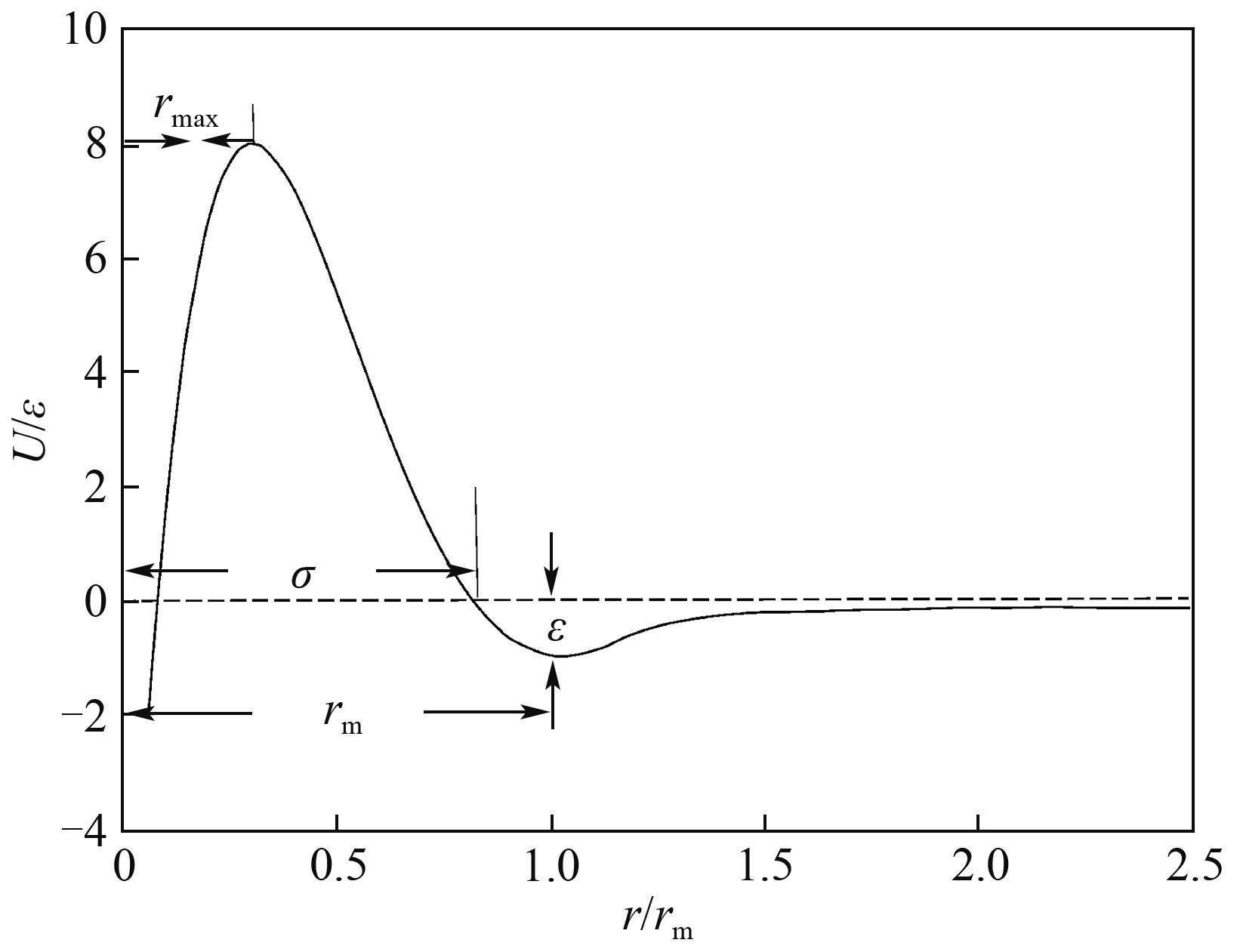
图4 Modified Buckingham(exp-6)势能曲线
Fig.4 Modified Buckingham (exp-6) potential curves

图5 UF6的rm (T)和ε(T)的温度依赖关系
Fig.5 Temperature dependence of rm(T) and ε(T) of UF6
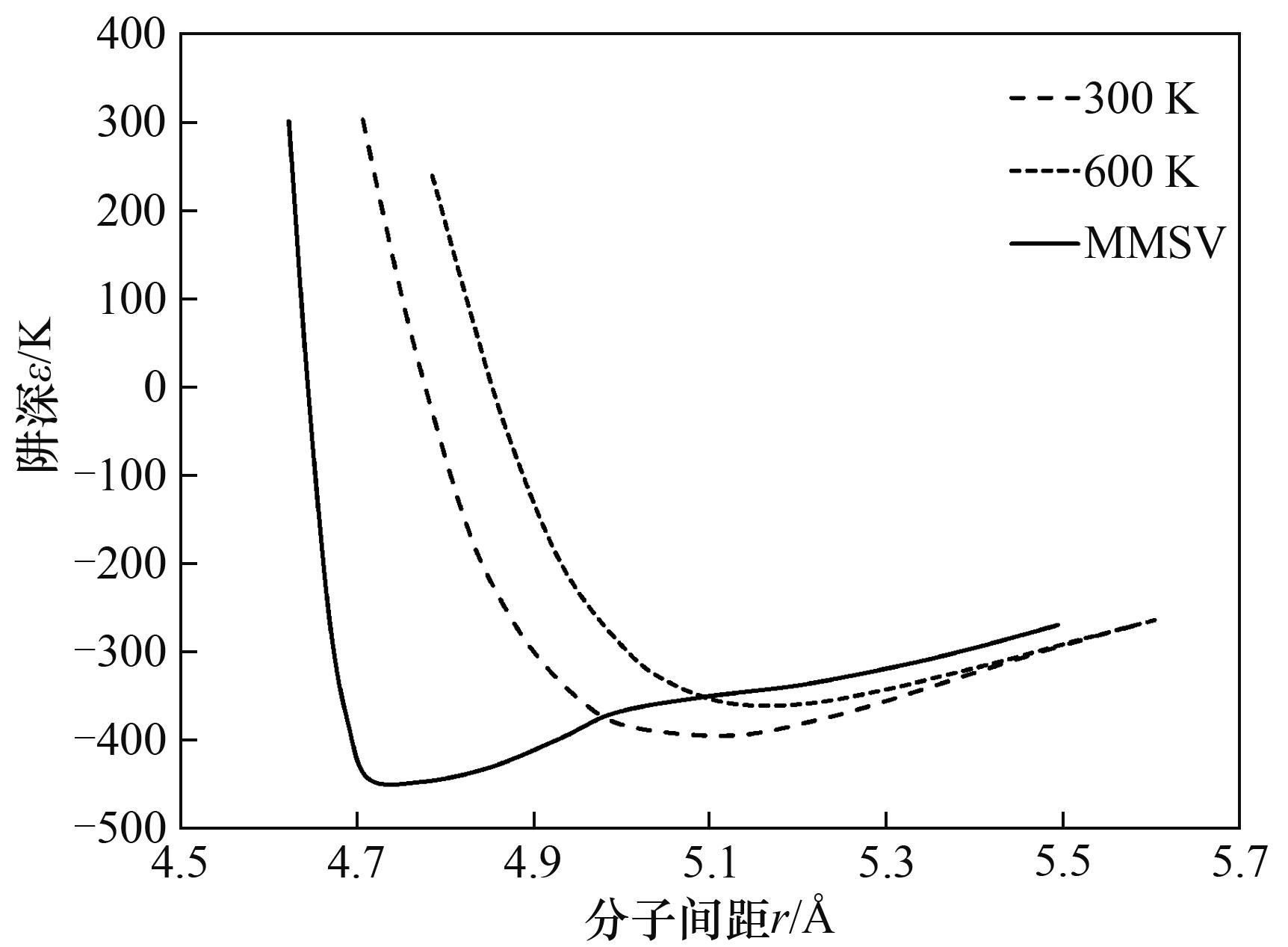
图6 TDIP与MMSV势能曲线的比较(SF6)[15]
Fig.6 Comparison of potential curves between TDIP and MMSV(SF6) [15]
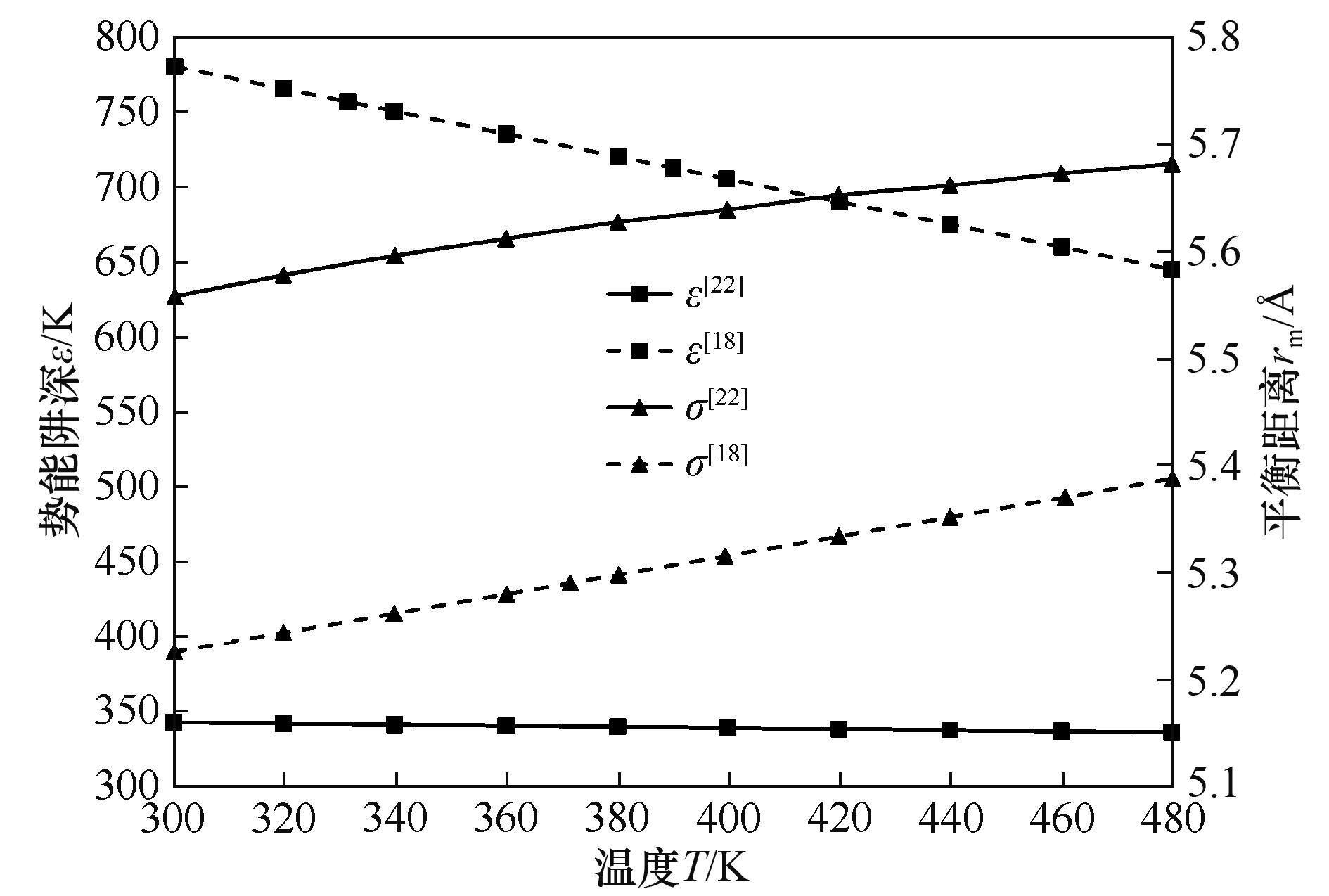
图7 温度对UF6碰撞直径(σ)和阱深(ε)的影响
Fig.7 Effect of temperature on UF6 collision diameter (σ) and well depth (ε)
| 方法 | 文献 | RMSD(B) |
|---|---|---|
| TDIP | [ | 35.449 |
| [ | 3.8931 | |
| TIIP | [ | 38.895 |
| [ | 151.31 | |
| [ | 147.66 | |
| [ | 261.01 | |
| [ | 11.187 |
表2 不同文献中UF6的第二维里系数的RMSD
Table 2 RMSD of second virial coefficient of UF6 in different literatures
| 方法 | 文献 | RMSD(B) |
|---|---|---|
| TDIP | [ | 35.449 |
| [ | 3.8931 | |
| TIIP | [ | 38.895 |
| [ | 151.31 | |
| [ | 147.66 | |
| [ | 261.01 | |
| [ | 11.187 |

图8 UF6的气液共存模拟曲线(TDIP和TIIP)与实验数据
Fig.8 Vapor-liquid coexistence curves (TDIP and TIIP) and experimental data of UF6
| 数据来源 | RMSD (B)/(cm3/mol) | |||
|---|---|---|---|---|
| Oh关联式 | Tsonopoulos 关联式 | Dymond关联式 | Zarkova & Hohm关联式 | |
| Ref.[ | 71 | 49.7 | 98.9 | 70.8 |
| Ref.[ | 77 | 94.3 | 40 | 77.3 |
| all data | 68 | 82.6 | 65.1 | 75.2 |
| 数据来源 | RMSD (η)/% | |||
| Oh关联式 | Lucas关联式 | Zarkova & Hohm关联式 | ||
| Ref.[ | 3.2 | 3 | 1.8 | |
| Ref.[ | 2.9 | 4.2 | 0.7 | |
| Ref.[ | 3.7 | 4.9 | 2.3 | |
| Ref.[ | 3.5 | 4.8 | 0.7 | |
| all data | 3.4 | 4.4 | 1.5 | |
表3 UF6第二维里系数和黏度的计算关联式与实验值的RMSD
Table 3 RMSD of calculation correlation and experimental data of second virial coefficient and viscosity of UF6
| 数据来源 | RMSD (B)/(cm3/mol) | |||
|---|---|---|---|---|
| Oh关联式 | Tsonopoulos 关联式 | Dymond关联式 | Zarkova & Hohm关联式 | |
| Ref.[ | 71 | 49.7 | 98.9 | 70.8 |
| Ref.[ | 77 | 94.3 | 40 | 77.3 |
| all data | 68 | 82.6 | 65.1 | 75.2 |
| 数据来源 | RMSD (η)/% | |||
| Oh关联式 | Lucas关联式 | Zarkova & Hohm关联式 | ||
| Ref.[ | 3.2 | 3 | 1.8 | |
| Ref.[ | 2.9 | 4.2 | 0.7 | |
| Ref.[ | 3.7 | 4.9 | 2.3 | |
| Ref.[ | 3.5 | 4.8 | 0.7 | |
| all data | 3.4 | 4.4 | 1.5 | |
| 1 | 邓茗文. 中国核电:硬核助力“双碳”目标 清洁赋能美好未来[J]. 可持续发展经济导刊, 2021(8): 44-48. |
| Deng M W. CNNP: developing the nuclear power to achieve carbon emission peaking and carbon-neutral target of China[J]. China Sustainability Tribune, 2021(8): 44-48. | |
| 2 | 吕程, 鞠吉, 曾爱武. 苯-噻吩-NMP三元体系汽液平衡的GEMC模拟[J]. 计算机与应用化学, 2016, 33(3): 309-312. |
| Lv C, Ju J, Zeng A W. Gemc simulation for benzene/thiophene/nmp ternary system[J]. Computers and Applied Chemistry, 2016, 33(3): 309-312. | |
| 3 | 吴东, 祁影霞, 杨喜. HFO-1234YF/HFC-32气-液相平衡特性研究[J]. 制冷技术, 2016, 36(1): 26-30, 34. |
| Wu D, Qi Y X, Yang X. Investigation on vapor-liquid equilibrium characteristics of HFO-1234YF/HFC-32[J]. Chinese Journal of Refrigeration Technology, 2016, 36(1): 26-30, 34. | |
| 4 | 董秀芹, 管肖肖, 马静. 二氧化碳-醋酸乙烯体系相平衡的GEMC模拟[J]. 计算机与应用化学, 2015, 32(10): 1182-1186. |
| Dong X Q, Guan X X, Ma J. Gibbs ensemble Monte Carlo simulations of binary mixture of CO2 and vinyl acetate[J]. Computers and Applied Chemistry, 2015, 32(10): 1182-1186. | |
| 5 | Potoff J J, Siepmann J I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen[J]. AIChE Journal, 2001, 47(7): 1676-1682. |
| 6 | Li H, Zhang J, Li D Y, et al. Monte Carlo simulations of vapour-liquid phase equilibrium and microstructure for the system containing azeotropes[J]. Molecular Simulation, 2017, 43(13/14/15/16): 1125-1133. |
| 7 | Li D Y, Gao Z Q, Vasudevan N K, et al. Molecular mechanism for azeotrope formation in ethanol/benzene binary mixtures through Gibbs ensemble Monte Carlo simulation[J]. The Journal of Physical Chemistry. B, 2020, 124(16): 3371-3386. |
| 8 | Malyshev V V. Equation of state for UF6 over the range of density variation to 0.0118 g/cm3 and temperature variation up to 367 K[J]. Atomic Energy (USSR), 1973, 34(1): 42-44. |
| 9 | Malyshev V V. Equations of state for UF6 for a wide range of state parameters[J]. Atomic Energy (USSR), 32(4):313-314. |
| 10 | Morizot P, Ostorero J, Plurien P. Viscosité et non-idéalité des hexafluorures de molybdène, de tungstène, d’uranium détermination de leurs paramètres moléculaires[J]. Journal De Chimie Physique, 1973, 70: 1582-1586. |
| 11 | Schneider B, Boring A M, Cohen J S. Interaction potentials for UF6 with itself and with rare-gas atoms[J]. Chemical Physics Letters, 1974, 27(4): 577-579. |
| 12 | Heintz A, Meisinger E, Lichtenthaler R N. Measured values of the second virial coefficient and determining the intermolecular interaction potential of gaseous uranium hexafluoride[J]. Berichte der Bunsengesellschaft fuer Physikalische Chemie, 1976, 80(2): 163-166. |
| 13 | Heintz A, Lichtenthaler R N. Data of the second virial coefficient of the gases PF5, MoF6, WF6 and JF5 and the determination of the intermolecular interaction potential[J]. Berichte Der Bunsengesellschaft/Physical Chemistry Chemical Physics, 1976, 80(10): 962-965. |
| 14 | Kirch P, Schütte R. Measurement of thermal diffusion and determination of the intermolecular potential of gaseous uranium hexafluoride[J]. The Journal of Chemical Physics, 1965, 42(10): 3729-3730. |
| 15 | Aziz R A, Slaman M J, Taylor W L, et al. An improved intermolecular potential for sulfur hexafluoride[J]. The Journal of Chemical Physics, 1991, 94(2): 1034-1038. |
| 16 | Coroiu I, Demco D E. Second virial coefficients and transport properties of hexafluoride gases from an improved intermolecular potential[J]. Zeitschrift Für Naturforschung A, 1997, 52(10): 748-756. |
| 17 | El-Kader M S A, Kalugina Y N. Dipole-octupole polarisability of uranium hexafluoride and theoretical prediction of anisotropic light-scattering spectrum using new intermolecular potential[J]. Molecular Physics, 2016, 114(1): 44-52. |
| 18 | Zarkova L, Pirgov P. Transport and equilibrium properties of UF6 gas simultaneously fitted by an effective isotropic potential with temperature-dependent parameters[J]. Journal of Physics B: Atomic, Molecular and Optical Physics, 1995, 28(19): 4269-4281. |
| 19 | Zarkova L. An isotropic intermolecular potential with temperature dependent effective parameters for heavy globular gases[J]. Molecular Physics, 1996, 88(2): 489-495. |
| 20 | Zarkova L, Hohm U. pVT-second virial coefficients B(T ), viscosity η(T ), and self-diffusion ρD(T) of the gases: BF3, CF4, SiF4, CCl4, SiCl4, SF6, MoF6, WF6, UF6, C(CH3)4, and Si(CH3)4 determined by means of an isotropic temperature-dependent potential[J]. Journal of Physical and Chemical Reference Data, 2002, 31(1): 183-216. |
| 21 | Zarkova L, Hohm U, Damyanova M. Viscosity and pVT-second virial coefficient of binary noble-globular gas and globular-globular gas mixtures calculated by means of an isotropic temperature-dependent potential[J]. Journal of Physical and Chemical Reference Data, 2003, 32(4): 1591-1705. |
| 22 | Al-Matar A K, Binous H. Vapor-liquid phase equilibrium diagram for uranium hexafluoride (UF6) using simplified temperature dependent intermolecular potential parameters (TDIP)[J]. Journal of Radioanalytical and Nuclear Chemistry, 2016, 310(1): 139-154. |
| 23 | Oh S K. Application of the group contribution concept to Kihara potential for estimating thermodynamic and transport properties(Ⅵ): Heavy globular molecules (SF6, MoF6, WF6, UF6, C(CH3)4, Si(CH3)4)[J]. Fluid Phase Equilibria, 2008, 271(1/2): 53-68. |
| 24 | Sadus R J. Molecular Simulation of Fluids[M]. Amsterdam:Elsevier, 2002. |
| 25 | Dove M T, Pawley G S. A molecular dynamics simulation study of the plastic crystalline phase of sulphur hexafluoride[J]. Journal of Physics C: Solid State Physics, 1983, 16(31): 5969-5983. |
| 26 | Lu H. Molecular dynamics simulations of solid sulphur hexafluoride[D]. Edinburgh: The University of Edinburgh, 1992. |
| 27 | 李洪, 张季, 李鑫钢, 等. 分子模拟方法计算相平衡热力学性质的研究进展[J]. 化工进展, 2017, 36(8): 2731-2741. |
| Li H, Zhang J, Li X G, et al. Progress in study on thermodynamic properties of phase equilibria using molecular simulation[J]. Chemical Industry and Engineering Progress, 2017, 36(8): 2731-2741. | |
| 28 | Rosenbluth M N, Rosenbluth A W. Monte Carlo calculation of the average extension of molecular chains[J]. The Journal of Chemical Physics, 1955, 23(2): 356-359. |
| 29 | Lísal M, Smith W R, Nezbeda I. Accurate vapour-liquid equilibrium calculations for complex systems using the reaction Gibbs ensemble Monte Carlo simulation method[J]. Fluid Phase Equilibria, 2001, 181(1/2): 127-146. |
| 30 | Lotfi A, Vrabec J, Fischer J. Vapour liquid equilibria of the Lennard-Jones fluid from the NpT plus test particle method[J]. Molecular Physics, 1992, 76(6): 1319-1333. |
| 31 | Yao J, Greenkorn R A, Chao K C. Monte Carlo simulation of the grand canonical ensemble[J]. Molecular Physics, 1982, 46(3): 587-594. |
| 32 | Kofke D A. Gibbs-Duhem integration: a new method for direct evaluation of phase coexistence by molecular simulation[J]. Molecular Physics, 1993, 78(6): 1331-1336. |
| 33 | Potoff J J, Panagiotopoulos A Z. Surface tension of the three-dimensional Lennard-Jones fluid from histogram-reweighting Monte Carlo simulations[J]. The Journal of Chemical Physics, 2000, 112(14): 6411-6415. |
| 34 | Boulougouris G C, Peristeras L D, Economou I G, et al. Predicting fluid phase equilibrium via histogram reweighting with Gibbs ensemble Monte Carlo simulations[J]. The Journal of Supercritical Fluids, 2010, 55(2): 503-509. |
| 35 | Ustinov E A, Do D D. Application of kinetic Monte Carlo method to equilibrium systems: vapour-liquid equilibria[J]. Journal of Colloid and Interface Science, 2012, 366(1): 216-223. |
| 36 | Nguyen V T, Tan S J, Do D D, et al. Application of kinetic Monte Carlo method to the vapour-liquid equilibria of associating fluids and their mixtures[J]. Molecular Simulation, 2016, 42(8): 642-654. |
| 37 | Fan C Y, Do D D, Nicholson D. New Monte Carlo simulation of adsorption of gases on surfaces and in pores: a concept of multibins[J]. The Journal of Physical Chemistry. B, 2011, 115(35): 10509-10517. |
| 38 | Fan C Y, Do D D, Nicholson D. A new and effective Bin-Monte Carlo scheme to study vapour-liquid equilibria and vapour-solid equilibria[J]. Fluid Phase Equilibria, 2012, 325: 53-65. |
| 39 | Jorgensen W L, Maxwell D S, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids[J]. Journal of the American Chemical Society, 1996, 118(45): 11225-11236. |
| 40 | Sun H. COMPASS: an ab initio force-field optimized for condensed-phase. Applications overview with details on alkane and benzene compounds[J]. The Journal of Physical Chemistry B, 1998, 102(38): 7338-7364. |
| 41 | Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields[J]. Journal of Computational Chemistry, 2010, 31(4): 671-690. |
| 42 | Martin M G, Siepmann J I. Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes[J]. The Journal of Physical Chemistry B, 1999, 103(21): 4508-4517. |
| 43 | Martin M G, Siepmann J I. Transferable potentials for phase equilibria. 1. United-atom description of n-alkanes[J]. The Journal of Physical Chemistry B, 1998, 102(14): 2569-2577. |
| 44 | Eggimann B L, Sunnarborg A J, Stern H D, et al. An online parameter and property database for the TraPPE force field[J]. Molecular Simulation, 2014, 40(1/2/3): 101-105. |
| 45 | Nath S K, Escobedo F A, de Pablo J J, et al. Simulation of vapor-liquid equilibria for alkane mixtures[J]. Industrial & Engineering Chemistry Research, 1998, 37(8): 3195-3202. |
| 46 | Marrink S J, de Vries A H, Mark A E. Coarse grained model for semiquantitative lipid simulations[J]. The Journal of Physical Chemistry B, 2004, 108(2): 750-760. |
| 47 | Shinoda W, DeVane R, Klein M L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants[J]. Molecular Simulation, 2007, 33(1/2): 27-36. |
| 48 | Olivet A, Vega L F. Optimized molecular force field for sulfur hexafluoride simulations[J]. The Journal of Chemical Physics, 2007, 126(14): 144502. |
| 49 | Lee K H, Lombardo M, Sandler S I. The generalized van der Waals partition function(Ⅱ): Application to the square-well fluid[J]. Fluid Phase Equilibria, 1985, 21(3): 177-196. |
| 50 | Stone A. The Theory of Intermolecular Forces[M]. Oxford: Oxford University Press, 2013. |
| 51 | Rice W E, Hirschfelder J O. Second virial coefficients of gases obeying a modified Buckingham (exp-six) potential[J]. The Journal of Chemical Physics, 1954, 22(2): 187-192. |
| 52 | Kihara T. Intermolecular Forces[M]. New York: John Wiley & Sons, 1978. |
| 53 | Parson J M, Siska P E, Lee Y T. Intermolecular potentials from crossed-beam differential elastic scattering measurements (Ⅳ): Ar+Ar[J]. The Journal of Chemical Physics, 1972, 56(4): 1511-1516. |
| 54 | Pack R T, Piper E, Pfeffer G A, et al. Multiproperty empirical anisotropic intermolecular potentials (Ⅱ): HeSF6 and NeSF6 [J]. The Journal of Chemical Physics, 1984, 80(10): 4940-4950. |
| 55 | Pack R T, Valentini J J, Becker C H, et al. Multiproperty empirical interatomic potentials for ArXe and KrXe[J]. The Journal of Chemical Physics, 1982, 77(11): 5475-5485. |
| 56 | Meng L, Duan Y Y. Site-site potential function and second virial coefficients for linear molecules[J]. Molecular Physics, 2006, 104(18): 2891-2899. |
| 57 | Zhang Y, Wang S, He M G. Modified 2CLJDQP model and the second virial coefficients for linear molecules[J]. Chinese Physics B, 2014, 23(12): 125101. |
| 58 | Meinander N. An isotropic intermolecular potential for sulfur hexafluoride based on the collision-induced light scattering spectrum, viscosity, and virial coefficient data[J]. The Journal of Chemical Physics, 1993, 99(11): 8654-8667. |
| 59 | Stefanov B, Zarkova L. The equilibrium and transport properties of heavy fluorine-containing gases predicted with the use of the vibrationally-excited-states-of-molecules (VESM) model[J]. High Temperatures-High Pressures, 1993, 25: 481-486. |
| 60 | Zarkova L, Pirgov P. The isotropic temperature-dependent potential describing the binary interactions in gaseous and[J]. Journal of Physics B: Atomic, Molecular and Optical Physics, 1996, 29(19): 4411-4422. |
| 61 | Stefanov B, Zarkova L. The model of vibrationally excited states of molecules as a tool for calculating thermodynamic and transport properties of molecular gases: SF6 as an example[J]. High Temperatures-High Pressures, 1993, 25: 487-490. |
| 62 | Zarkova L, Pirgov P. Thermophysical properties of diluted F-containing heavy globular gases predicted by means of temperature dependent effective isotropic potential[J]. Vacuum, 1997, 48(1): 21-27. |
| 63 | Zarkova L, Hohm U. Effective (n-6) Lennard-Jones potentials with temperature-dependent parameters introduced for accurate calculation of equilibrium and transport properties of ethene, propene, butene, and cyclopropane[J]. Journal of Chemical & Engineering Data, 2009, 54(6): 1648-1655. |
| 64 | Dymond J H, Marsh K N, Wilhoit R C, et al. Virial Coefficients of Pure Gases[M]. Landord-Bornstein: Springer, 2003. |
| 65 | DeWitt R. Uranium Hexafluoride: A Survey of The Physicochemical Properties[R]. Office of Scientific and Technical Information (OSTI), 1960. |
| 66 | Verkhivker G P, Tetel’baum S D, Konyaeva G P. Thermodynamic properties of uranium hexafluoride (UF6)[J]. Soviet Atomic Energy, 1968, 24(2): 191-195. |
| 67 | Malyshev V V. Kihara interaction potentials for octahedral molecular systems SF6, MoF6, WF6, and UF6 [J]. High Temperature, 1974, 12(5): 979-983. |
| 68 | Hellemans J M, Kestin J, Ro S T. The viscosity of CH4, CF4 and SF6 over a range of temperatures[J]. Physica, 1973, 65(2): 376-380. |
| 69 | Kestin J, Khalifa H E, Ro S T, et al. The viscosity and diffusion coefficients of eighteen binary gaseous systems[J]. Physica A: Statistical Mechanics and Its Applications, 1977, 88(2): 242-260. |
| 70 | Strehlow T, Vogel E. Temperature dependence and initial density dependence of the viscosity of sulphur hexafluoride[J]. Physica A: Statistical Mechanics and Its Applications, 1989, 161(1): 101-117. |
| 71 | Kestin J, Ro S T, Wakeham W A. Reference values of the viscosity of twelve gases at 25℃[J]. Trans. Faraday Soc., 1971, 67: 2308-2313. |
| 72 | Kestin J, Khalifa H E, Wakeham W A. Viscosity of multicomponent mixtures of four complex gases[J]. The Journal of Chemical Physics, 1976, 65(12): 5186-5188. |
| 73 | Kestin J, Khalifa H E, Wakeham W A. The viscosity of gaseous mixtures containing krypton[J]. The Journal of Chemical Physics, 1977, 67(9): 4254-4259. |
| 74 | Abe Y, Kestin J, Khalifa H E, et al. The viscosity and diffusion coefficients of the mixtures of light hydrocarbons with other polyatomic gases[J]. Berichte Der Bunsengesellschaft Für Physikalische Chemie, 1979, 83(3): 271-276. |
| 75 | Hoogland J H B, van den Berg H R, Trappeniers N J. Measurements of the viscosity of sulfur hexaflouride up to 100 bar by a capillary-flow viscometer[J]. Physica A: Statistical Mechanics and Its Applications, 1985, 134(1): 169-192. |
| 76 | Boushehri A, Bzowski J, Kestin J, et al. Equilibrium and transport properties of eleven polyatomic gases at low density[J]. Journal of Physical and Chemical Reference Data, 1987, 16(3): 445-466. |
| 77 | Trengove R D, Wakeham W A. The viscosity of carbon dioxide, methane, and sulfur hexafluoride in the limit of zero density[J]. Journal of Physical and Chemical Reference Data, 1987, 16(2): 175-187. |
| 78 | Lichtenthaler R N, Schafer K. Intermolecular forces of spherical and non-spherical molecules calculated from second virial coefficients[J]. Berichte der Bunsen-Gesellschaft fur Physikalische Chemie, 1969, 73(1): 42-+. |
| 79 | Bzowski J, Kestin J, Mason E A, et al. Equilibrium and transport properties of gas mixtures at low density: eleven polyatomic gases and five noble gases[J]. Journal of Physical and Chemical Reference Data, 1990, 19(5): 1179-1232. |
| 80 | Zarkova L, Hohm U, Damyanova M. Comparison of Lorentz-Berthelot and Tang-Toennies mixing rules using an isotropic temperature-dependent potential applied to the thermophysical properties of binary gas mixtures of CH4, CF4, SF6, and C(CH3)4 with Ar, Kr, and Xe[J]. International Journal of Thermophysics, 2004, 25(6): 1775-1798. |
| 81 | Tsonopoulos C. An empirical correlation of second virial coefficients[J]. AIChE Journal, 1974, 20(2): 263-272. |
| 82 | Lucas K. Phase Equilibria and Fluid Properties in the Chemical Industry[M]. Frankfurt: Dechema, 1980: 100. |
| 83 | Kigoshi K. On the viscosity of the uranium hexafluoride[J]. Bulletin of the Chemical Society of Japan, 1950, 23(2): 67-68. |
| 84 | Myerson A L, Eicher J H. The viscosity of gaseous uranium Hexafluoride1[J]. Journal of the American Chemical Society, 1952, 74(11): 2758-2761. |
| 85 | Llewellyn D R. 5. Some physical properties of uranium hexafluoride[J]. Journal of the Chemical Society (Resumed), 1953: 28. |
| [1] | 宋明昊, 赵霏, 刘淑晴, 李国选, 杨声, 雷志刚. 离子液体脱除模拟油中挥发酚的多尺度模拟与研究[J]. 化工学报, 2023, 74(9): 3654-3664. |
| [2] | 胡建波, 刘洪超, 胡齐, 黄美英, 宋先雨, 赵双良. 有机笼跨细胞膜易位行为的分子动力学模拟研究[J]. 化工学报, 2023, 74(9): 3756-3765. |
| [3] | 赵佳佳, 田世祥, 李鹏, 谢洪高. SiO2-H2O纳米流体强化煤尘润湿性的微观机理研究[J]. 化工学报, 2023, 74(9): 3931-3945. |
| [4] | 汪林正, 陆俞冰, 张睿智, 罗永浩. 基于分子动力学模拟的VOCs热氧化特性分析[J]. 化工学报, 2023, 74(8): 3242-3255. |
| [5] | 陈吉, 洪泽, 雷昭, 凌强, 赵志刚, 彭陈辉, 崔平. 基于分子动力学的焦炭溶损反应及其机理研究[J]. 化工学报, 2023, 74(7): 2935-2946. |
| [6] | 董明, 徐进良, 刘广林. 超临界水非均质特性分子动力学研究[J]. 化工学报, 2023, 74(7): 2836-2847. |
| [7] | 刘远超, 蒋旭浩, 邵钶, 徐一帆, 钟建斌, 李耑. 几何尺寸及缺陷对石墨炔纳米带热输运特性的影响[J]. 化工学报, 2023, 74(6): 2708-2716. |
| [8] | 姚晓宇, 沈俊, 李健, 李振兴, 康慧芳, 唐博, 董学强, 公茂琼. 流体气液临界参数测量方法研究进展[J]. 化工学报, 2023, 74(5): 1847-1861. |
| [9] | 陈科, 杜理, 曾英, 任思颖, 于旭东. 四元体系LiCl+MgCl2+CaCl2+H2O 323.2 K相平衡研究及计算[J]. 化工学报, 2023, 74(5): 1896-1903. |
| [10] | 顾浩, 张福建, 刘珍, 周文轩, 张鹏, 张忠强. 力电耦合作用下多孔石墨烯膜时间维度的脱盐性能及机理研究[J]. 化工学报, 2023, 74(5): 2067-2074. |
| [11] | 李辰鑫, 潘艳秋, 何流, 牛亚宾, 俞路. 基于碳微晶结构的炭膜模型及其气体分离模拟[J]. 化工学报, 2023, 74(5): 2057-2066. |
| [12] | 毛元敬, 杨智, 莫松平, 郭浩, 陈颖, 罗向龙, 陈健勇, 梁颖宗. C6~C10烷醇的SAFT-VR Mie状态方程参数回归及其热物性研究[J]. 化工学报, 2023, 74(3): 1033-1041. |
| [13] | 程文婷, 李杰, 徐丽, 程芳琴, 刘国际. AlCl3·6H2O在FeCl3、CaCl2、KCl及KCl–FeCl3溶液中溶解度的实验及预测[J]. 化工学报, 2023, 74(2): 642-652. |
| [14] | 廖艺, 牛亚宾, 潘艳秋, 俞路. 复配表面活性剂对油水界面行为和性质影响的模拟研究[J]. 化工学报, 2022, 73(9): 4003-4014. |
| [15] | 孙哲, 金华强, 李康, 顾江萍, 黄跃进, 沈希. 基于知识数据化表达的制冷空调系统故障诊断方法[J]. 化工学报, 2022, 73(7): 3131-3144. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
 京公网安备 11010102001995号
京公网安备 11010102001995号

