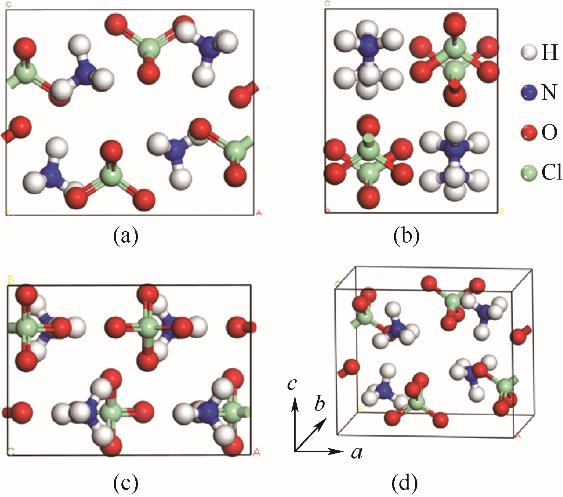
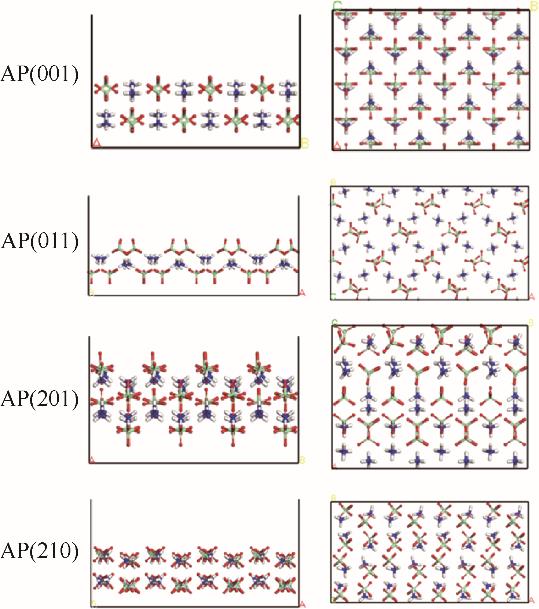
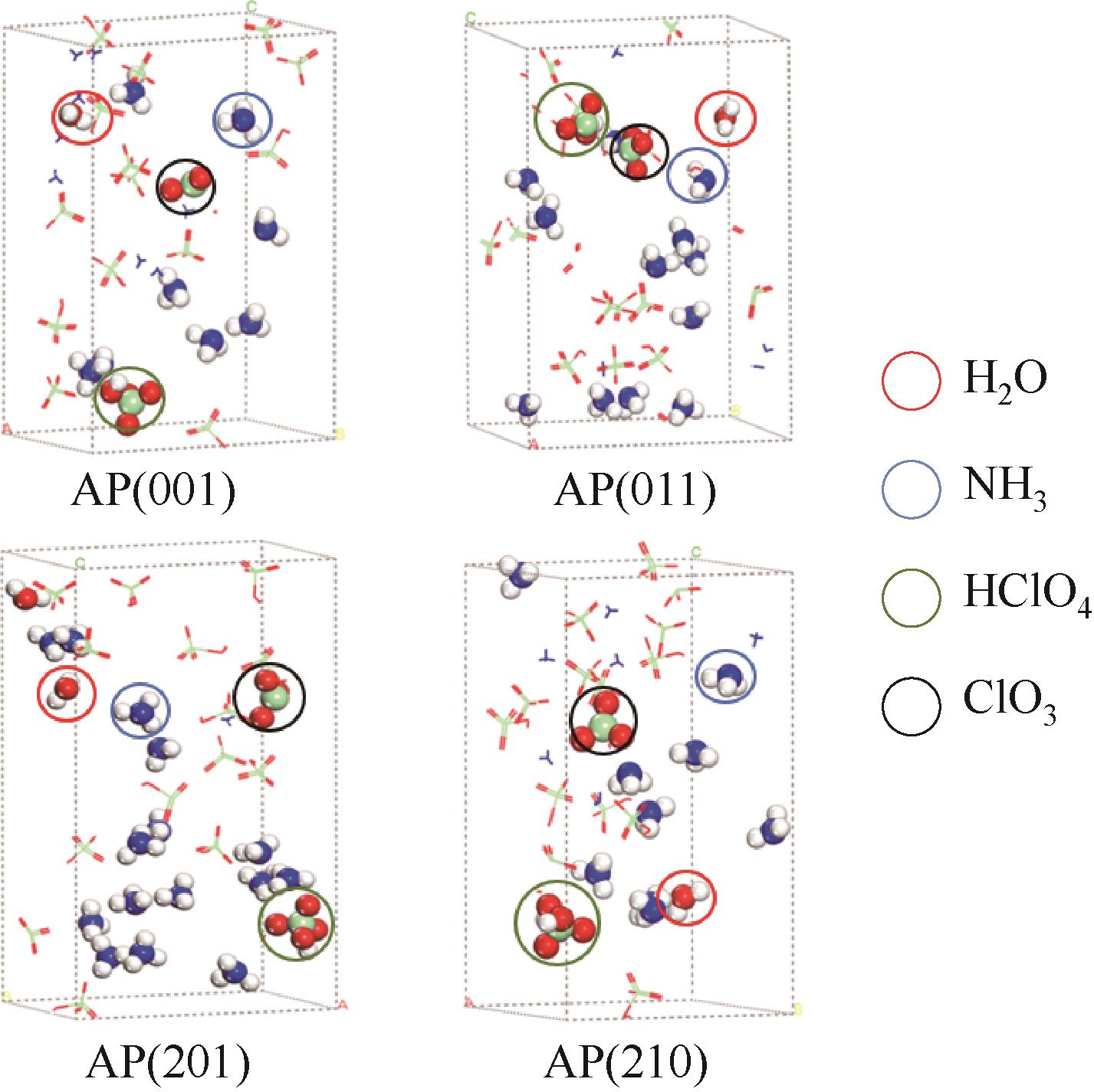
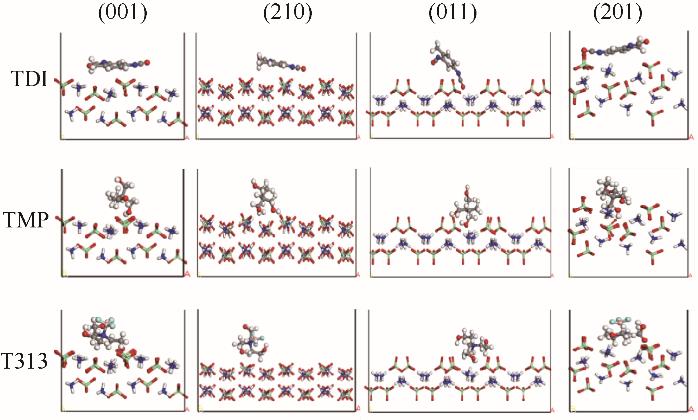
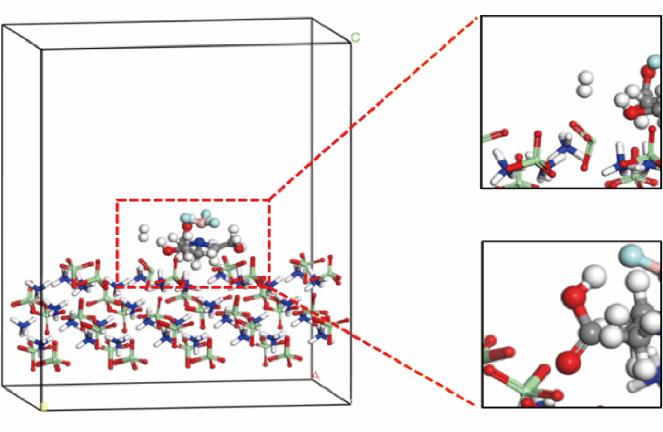
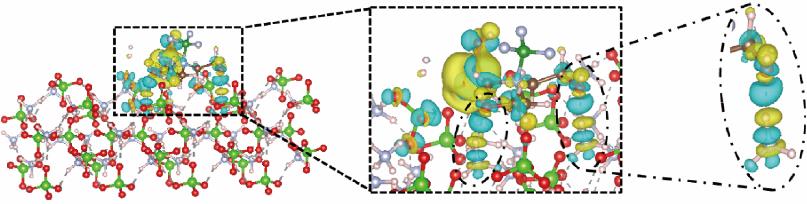
| 1 |
Chaturvedi S, Dave P N. Solid propellants: AP/HTPB composite propellants[J]. Arabian Journal of Chemistry, 2019, 12(8): 2061-2068.
|
| 2 |
Hunter S, Davidson A J, Morrison C A, et al. Combined experimental and computational hydrostatic compression study of crystalline ammonium perchlorate[J]. The Journal of Physical Chemistry C, 2011, 115(38): 18782-18788.
|
| 3 |
Kellogg R, Lapidus S, Hedman T, et al. Synchrotron based measurement of the temperature dependent thermal expansion coefficient of ammonium perchlorate[J]. Propellants, Explosives, Pyrotechnics, 2020, 45(3): 480-485.
|
| 4 |
Kroonblawd M P, Koroglu B, Zaug J M, et al. Effects of pressure on the structure and lattice dynamics of ammonium perchlorate: a combined experimental and theoretical study[J]. The Journal of Chemical Physics, 2018, 149(3): 034501.
|
| 5 |
Zhu W H, Wei T, Zhu W, et al. Comparative DFT study of crystalline ammonium perchlorate and ammonium dinitramide[J]. The Journal of Physical Chemistry. A, 2008, 112(20): 4688-4693.
|
| 6 |
Liu Y B, Zhang S H, Gou R J, et al. Theoretical study on the intermolecular interactions between energetic oxidizer and pyrazine - 1, 4 - dioxide[J]. Materials Today Communications, 2020, 24: 101020.
|
| 7 |
Wu Q, Li C L, Tan L H, et al. Comparative DFT and DFT-D studies on structural, electronic, vibrational and absorption properties of crystalline ammonium perchlorate[J]. RSC Advances, 2016, 6(54): 48489-48497.
|
| 8 |
Yedukondalu N, Vaitheeswaran G. Polymorphism, phase transition, and lattice dynamics of energetic oxidizer ammonium perchlorate under high pressure[J]. The Journal of Physical Chemistry C, 2019, 123(4): 2114-2126.
|
| 9 |
Yeh I C, Andzelm J W. Computational study of structural and energetic properties of ammonium perchlorate at interfaces[J]. The Journal of Physical Chemistry C, 2021, 125(22): 12297-12304.
|
| 10 |
Forquet V, Miró Sabaté C, Chermette H, et al. Energetic properties of rocket propellants evaluated through the computational determination of heats of formation of nitrogen-rich compounds[J]. Chemistry-An Asian Journal, 2016, 11(5): 730-744.
|
| 11 |
Tang P F, Yang B, Li R, et al. Ti3C2 MXene: a reactive combustion catalyst for efficient burning rate control of ammonium perchlorate based solid propellant[J]. Carbon, 2022, 186: 678-687.
|
| 12 |
吴芳, 熊中年, 燕为光, 等. Bu-NENA/PBT推进剂安全性能[J]. 固体火箭技术, 2019, 42(4): 483-487.
|
|
Wu F, Xiong Z N, Yan W G, et al. Safety properties of Bu-NENA/PBT propellants[J]. Journal of Solid Rocket Technology, 2019, 42(4): 483-487.
|
| 13 |
周水平, 王艳萍, 唐根, 等. 黏合剂固化网络参数对叠氮聚醚推进剂低温性能的影响[J]. 化学推进剂与高分子材料, 2017, 15(5): 49-54.
|
|
Zhou S P, Wang Y P, Tang G, et al. Influence of binder curing network parameters on cryogenic performance of azido polyether propellant[J]. Chemical Propellants & Polymeric Materials, 2017, 15(5): 49-54.
|
| 14 |
周水平, 吴芳, 唐根, 等. 含能交联剂对PBT高能推进剂力学性能的影响[J]. 化学推进剂与高分子材料, 2016, 14(4): 54-59.
|
|
Zhou S P, Wu F, Tang G, et al. Influence of energetic crosslinking agents on mechanical performance of PBT-based high energy propellants[J]. Chemical Propellants & Polymeric Materials, 2016, 14(4): 54-59.
|
| 15 |
姚维尚, 李倩, 谭惠民. NEPE推进剂黏合剂性能的分子模拟研究[J]. 含能材料, 2007, 15(6): 650-655.
|
|
Yao W S, Li Q, Tan H M. Molecular simulation on properties of NEPE propellant binders[J]. Chinese Journal of Energetic Materials, 2007, 15(6): 650-655.
|
| 16 |
焦东明, 杨月诚, 强洪夫, 等. 键合剂对HTPB与Al/Al2O3之间界面作用的分子模拟[J]. 火炸药学报, 2009, 32(4): 60-63.
|
|
Jiao D M, Yang Y C, Qiang H F, et al. Molecular simulation of effect of bonding agents on interface interaction for HTPB and Al/Al2O3 [J]. Chinese Journal of Explosives & Propellants, 2009, 32(4): 60-63.
|
| 17 |
Choi C S, Prask H J, Prince E. Crystal structure of NH4ClO4 at 298, 78, and 10 by neutron diffraction[J]. The Journal of Chemical Physics, 1974, 61(9): 3523-3529.
|
| 18 |
Mao Y, Hu P. Identification of the active sites and mechanism for partial methane oxidation to methanol over copper-exchanged CHA zeolites[J]. Science China Chemistry, 2020, 63(6): 850-859.
|
| 19 |
Wei Z Z, Yao Z H, Zhou Q, et al. Optimizing alkyne hydrogenation performance of Pd on carbon in situ decorated with oxygen-deficient TiO2 by integrating the reaction and diffusion[J]. ACS Catalysis, 2019, 9(12): 10656-10667.
|
| 20 |
Yao Z H, Guo C X, Mao Y, et al. Quantitative determination of C—C coupling mechanisms and detailed analyses on the activity and selectivity for Fischer-Tropsch synthesis on Co(0001): microkinetic modeling with coverage effects[J]. ACS Catalysis, 2019, 9(7): 5957-5973.
|
| 21 |
Yao Z H, Zhao J Y, Bunting R J, et al. Quantitative insights into the reaction mechanism for the direct synthesis of H2O2 over transition metals: coverage-dependent microkinetic modeling[J]. ACS Catalysis, 2021, 11(3): 1202-1221.
|
| 22 |
Yao Z H, Zhao J Y, Zhao C X, et al. A first-principles study of reaction mechanism over carbon decorated oxygen-deficient TiO2 supported Pd catalyst in direct synthesis of H2O2 [J]. Chinese Journal of Chemical Engineering, 2021, 31: 126-134.
|
| 23 |
Khan M A S, Vijayalakshmi R, Singh A, et al. Morphology of ammonium perchlorate in the presence of ethylene glycol as an additive: a first principle study[J]. CrystEngComm, 2019, 21(48): 7519-7527.
|
| 24 |
Kang L, Li S R, Wang B, et al. Exploration of the energetic material ammonium perchlorate at high pressures: combined Raman spectroscopy and X-ray diffraction study[J]. The Journal of Physical Chemistry C, 2018, 122(28): 15937-15944.
|
| 25 |
Ma Z Y, Pang A M, Li W, et al. Preparation and characterization of ultra-fine ammonium perchlorate crystals using a microfluidic system combined with ultrasonication[J]. Chemical Engineering Journal, 2021, 405: 126516.
|
| 26 |
Xie P F, Ding J, Yao Z H, et al. Oxo dicopper anchored on carbon nitride for selective oxidation of methane[J]. Nature Communications, 2022, 13(1): 1375.
|
| 27 |
Reuter K, Scheffler M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure[J]. Physical Review B, 2001, 65(3): 035406.
|
| 28 |
Reuter K, Scheffler M. Composition and structure of the RuO2(110) surface in an O2 and CO environment: implications for the catalytic formation of CO2 [J]. Physical Review B, 2003, 68(4): 045407.
|
| 29 |
Reuter K, Scheffler M. Oxide formation at the surface of late 4d transition metals: insights from first-principles atomistic thermodynamics[J]. Applied Physics A, 2004, 78(6): 793-798.
|
| 30 |
Rogal J, Reuter K, Scheffler M. Thermodynamic stability of PdO surfaces[J]. Physical Review B, 2004, 69(7): 075421.
|
| 31 |
Therrien A J, Zhang R Q, Lucci F R, et al. Structurally accurate model for the “29”-structure of Cu x O/Cu(111): a DFT and STM study[J]. The Journal of Physical Chemistry C, 2016, 120(20): 10879-10886.
|
 ), Ge FENG1, Jinyan ZHAO1, Jiayuan LI1, Shengwei DENG1, Jingnan ZHENG1, Wenwen LI1, Yaqiu WANG1, Lan SHEN1, Xu LIU1, Weiwei XU1, Jianguo WANG1, Shibin WANG1, Zihao YAO1(
), Ge FENG1, Jinyan ZHAO1, Jiayuan LI1, Shengwei DENG1, Jingnan ZHENG1, Wenwen LI1, Yaqiu WANG1, Lan SHEN1, Xu LIU1, Weiwei XU1, Jianguo WANG1, Shibin WANG1, Zihao YAO1(