CIESC Journal ›› 2019, Vol. 70 ›› Issue (6): 2051-2059.DOI: 10.11949/0438-1157.20181187
• Reviews and monographs • Previous Articles Next Articles
Liubin SONG( ),Anxian LI,Zhongliang XIAO(),Zhenzhen CHI,Zhong CAO
),Anxian LI,Zhongliang XIAO(),Zhenzhen CHI,Zhong CAO
Received:2018-10-12
Revised:2019-03-26
Online:2019-06-05
Published:2019-06-05
Contact:
Zhongliang XIAO
宋刘斌(),黎安娴,肖忠良(),池振振,曹忠
通讯作者:
肖忠良
作者简介:<named-content content-type="corresp-name">宋刘斌</named-content>(1981—),男,博士,讲师,<email>kjcsongliubin@163.com</email>
基金资助:CLC Number:
Liubin SONG, Anxian LI, Zhongliang XIAO, Zhenzhen CHI, Zhong CAO. Application research status of first-principles in lithium-ion battery electrode materials[J]. CIESC Journal, 2019, 70(6): 2051-2059.
宋刘斌, 黎安娴, 肖忠良, 池振振, 曹忠. 第一性原理在锂离子电池电极材料中的应用研究[J]. 化工学报, 2019, 70(6): 2051-2059.
Add to citation manager EndNote|Ris|BibTeX

Fig.1 Applications of first principles calculations in electrode materials
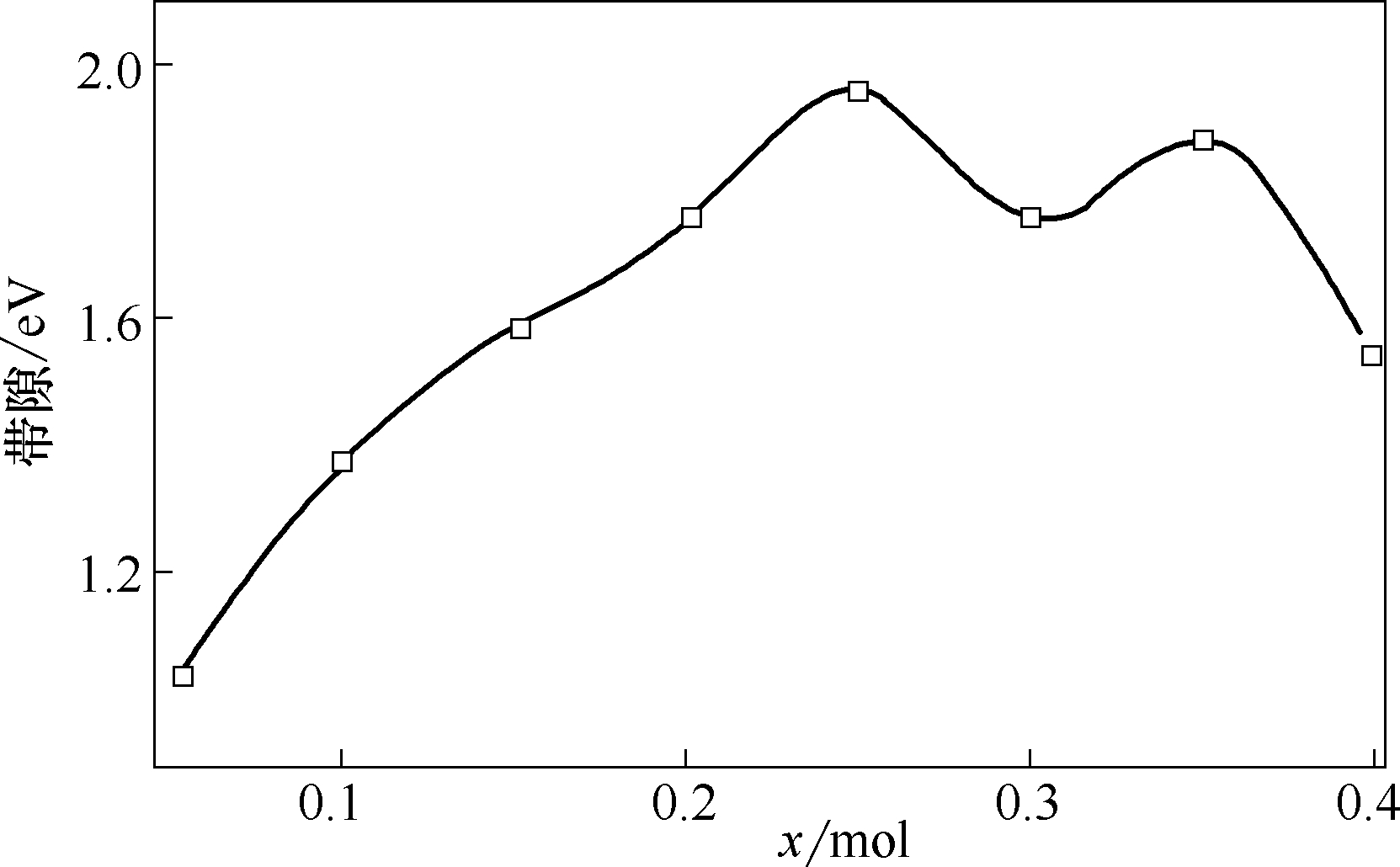
Fig.2 Band gap of materials with different doping amount x of Co[51]

Fig.3 Li diffusion profile for undoped and Al-doped NCM-523 using PBE[52]
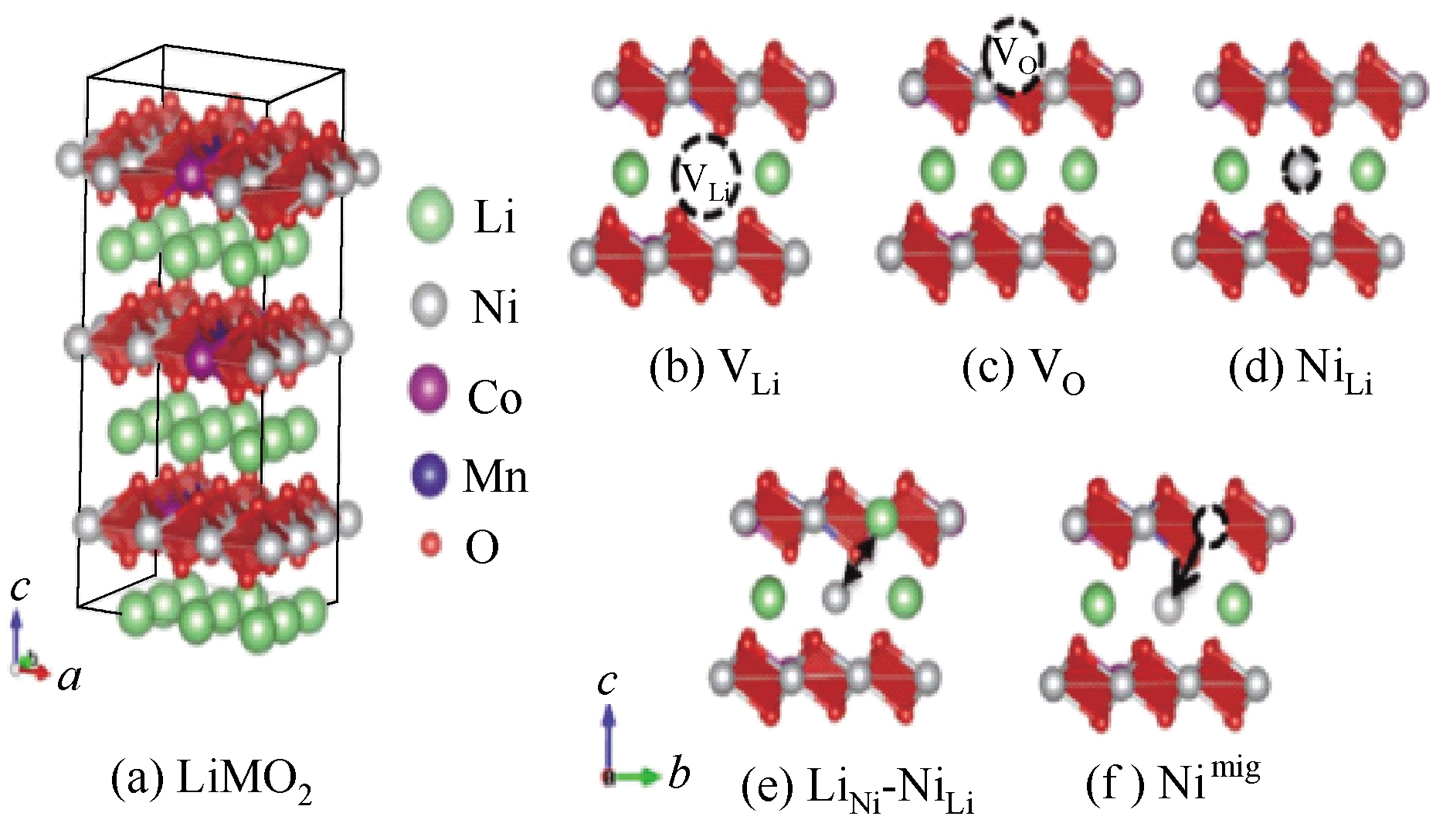
Fig.4 Atomic configurations for structure of LiMO2(a), where M= transition metals (Ni, Co,and Mn), and schematic structures for each of defects, Li vacancies (VLi)(b), oxygen vacancy(VO)(c), excess Ni (NiLi)(d), Li/Ni exchange (LiNi-NiLi)(e), and Ni migration (Nimig)(f)[63]
| 1 | 田萌 . 锂离子电池新型正极材料的第一性原理研究[D]. 北京: 中国科学院大学(中国科学院物理研究所), 2017. |
| Tian M . First-principles calculations on new cathode materials for lithium-ion batteries[D]. Beijing: The University of Chinese Academy of Sciences (Institute of Physics Chinese Academy of Sciences), 2017. | |
| 2 | Liu E , Wang J , Shi C , et al . Anomalous interfacial lithium storage in graphene/TiO2 for lithium ion batteries[J]. ACS Applied Materials & Interfaces, 2014, 6(20): 18147-18151. |
| 3 | Fei Z , Cococcioni M , Kang K , et al . The Li intercalation potential of LiMPO4 and LiMSiO4 olivines with M = Fe, Mn, Co, Ni[J]. Electrochemistry Communications, 2004, 6(11): 1144-1148. |
| 4 | 陈昌国, 刘艳, 司玉军, 等 . 锂离子电池正极材料LiFePO4的密度泛函研究[J]. 分子科学学报, 2007, 23(4): 248-252. |
| Chen C G , Liu Y , Si Y J , et al . Density functional study of cathode material LiFePO4 for lithium ion battery[J]. Journal of Molecular Science, 2007, 23(4): 248-252. | |
| 5 | Hassan A S , Navulla A , Meda L , et al . Molecular mechanisms for the lithiation of ruthenium oxide nanoplates as lithium-ion battery anode materials: an experimentally motivated computational study[J]. Journal of Physical Chemistry C, 2015, 119(18): 9705-9713. |
| 6 | Lv Y , Chen B , Zhao N , et al . Interfacial effect on the electrochemical properties of the layered graphene/metal sulfide composites as anode materials for Li-ion batteries[J]. Surface Science, 2016, 651: 10-15. |
| 7 | Iddir H , Benedek R . First-principles analysis of phase stability in layered–layered composite cathodes for lithium-ion batteries[J]. Chemistry of Materials, 2014, 26(7): 2407-2413. |
| 8 | Gao Y , Wang X , Ma J , et al . Selecting substituent elements for Li-rich Mn-based cathode materials by density functional theory (DFT) calculations[J]. Chemistry of Materials, 2015, 27(9): 3456-3461. |
| 9 | Wang T , Zhao N , Shi C , et al . Interface and doping effects on Li ion storage behavior of graphene/Li2O[J]. Journal of Physical Chemistry C, 2017, 121(36): 19559-19567. |
| 10 | 薛雷, 张秀娟, 张淑凯 . 锂离子电池层状正极材料第一性原理计算新进展[J]. 材料导报, 2016, 30(1): 122-127. |
| Xue L , Zhang X J , Zhang S K . New progresses of first-principles calculation on layered cathode materials for lithium ion batteries[J]. Material Review, 2016, 30(1): 122-127. | |
| 11 | 沈丁, 李犇, 杨绍斌, 等 . 锂离子电池聚阴离子型正极材料的第一性原理研究进展[J]. 化工进展, 2013, 32(4): 837-841. |
| Shen D , Li B , Yang S B , et al . Research progress of first principle of polyanion type cathode material for lithium-ion battery[J]. Chemical Industry and Engineering Progress, 2013, 32(4): 837-841. | |
| 12 | 徐宇虹, 尹鸽平, 左朋建 . 锂离子电池正极材料的第一性原理[J]. 化学进展, 2008, 20(11): 1827-1833. |
| Xu Y H , Yin G P , Zuo P J . First principle calculations of cathode in Li-ion batteries[J]. Chemical Industry and Engineering Progress, 2008, 20(11): 1827-1833. | |
| 13 | 张培新, 陈建华, 魏群 . 掺杂材料分子模拟与计算[M]. 北京: 科学出版社, 2012. |
| Zhang P X , Chen J H , Wei Q . Molecular Simulation and Calculation of Doping Materials[M]. Beijing: Science Press, 2012. | |
| 14 | 胡英, 刘洪来 . 密度泛函理论[M]. 北京: 科学出版社, 2016. |
| Hu Y, Liu H L, Density Functional Theory[M]. Beijing: Science Press, 2016. | |
| 15 | Hohenberg P , Kohn W . Inhomogeneous electron gas[J]. Phys. Rev., 1964, 136(3B): B864–B871. |
| 16 | 林梦海 . 量子化学计算方法与应用[M]. 北京: 科学出版社, 2005. |
| Lin M H . Quantum Chemistry Calculation Methods and Applications[M]. Beijing: Science Press, 2005. | |
| 17 | Kohn W , Sham L J . Self-consistent equations including exchange and correlation effects[J]. Physical Review, 2008, 140(4A): A1133-A1138. |
| 18 | Perdew J P , Burke K , Ernzerhof M . Generalized gradient approximation made simple[J]. Phys. Rev. Lett., 1996, 77: 3865. |
| 19 | Perdew J P , Wang Y . Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B Condensed Matter, 1992, 45(23): 13244. |
| 20 | 宋刘斌 . 锂离子电池的热电化学研究及其电极材料的计算与模拟[D]. 长沙: 中南大学, 2013. |
| Song L B . The thermo-eletrochemical study on lithium ion batteries and numerical calculation and simulation of electrode material in lithium ion batteries[D]. Changsha: Central South University, 2013. | |
| 21 | Kresse G , Furthmüller J . Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Phys. Rev. B Condens Matter, 1996, 54(16): 11169-11186. |
| 22 | Kresse G , Joubert D . From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Phys. Rev. B, 1999, 59(3): 1758-1775. |
| 23 | Blöchl P . Projector augmented-wave method[J]. Phys. Rev. B Condens Matter, 1994, 50(24):17953-17979. |
| 24 | 孙超 . 基于密度泛函理论的材料设计:VO2相变温度的调控和LiFePO4电导率的提高[D]. 上海: 上海大学, 2015. |
| Sun C . Materials design based on density functional theory: modulation of transition temperature of VO2 and improvement of conductivity of LiFePO4 [D]. Shanghai: Shanghai University, 2015. | |
| 25 | Payne M C , Teter M P , Allan D C , et al . Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients[J]. Reviews of Modern Physics(United States), 1992, 64(4): 1045-1097. |
| 26 | 孔婷婷 . 钛锂铝类水滑石/炭复合材料的制备及CO2吸附与光催化研究[D]. 西安: 西安科技大学, 2017. |
| Kong T T . Preparation and CO2 adsorption, photocatalysis performance of Ti/Li/Al-LDHs/coke composite[D]. Xi an: Xi an University of Science and Technology, 2017. | |
| 27 | 张福州 . 广义层错能的第一性原理计算及定域性分析[D]. 重庆: 重庆大学, 2008. |
| Zhang F Z . Generalized stacking fault energy calculation from first-principles and analysis of local approximation[D]. Chongqing: Chongqing University, 2008. | |
| 28 | 温亚娟 . 典型热电材料的第一原理计算[D]. 杭州: 杭州电子科技大学, 2014. |
| Wen Y J . The first principle studies of several typical thermoelectric materials[D]. Hangzhou: Hangzhou Dianzi University, 2014. | |
| 29 | Nosengo N . The material code[J]. Nature, 2016, 533(7602): 22. |
| 30 | Umebayashi Y , Mitsugi T , Fukuda S , et al . Lithium ion solvation in room-temperature ionic liquids involving bis(trifluoromethanesulfonyl) imide anion studied by Raman spectroscopy and DFT calculations[J]. Journal of Physical Chemistry B, 2007, 111(45): 13028. |
| 31 | Nakayama M , Yamada S , Jalem R , et al . Density functional studies of olivine-type LiFePO4, and NaFePO4, as positive electrode materials for rechargeable lithium and sodium ion batteries[J]. Solid State Ionics, 2016, 286: 40-44. |
| 32 | Xiao R , Li H , Chen L . Density functional investigation on Li2MnO3 [J]. Chemistry of Materials, 2012, 24(21): 4242-4251. |
| 33 | Anisimov V I . First-principles calculations of the electronic structure and spectra of strongly correlated systems: LDA+U method[J]. Journal of Physics Condensed Matter, 1995, 9(35): 7359-7367. |
| 34 | Kong F , Longo R C , Min S P , et al . Ab initio study of doping effects on LiMnO2 and Li2MnO3 cathode materials for Li-ion batteries[J]. Journal of Materials Chemistry A, 2015, 3(16): 8489-8500. |
| 35 | Reimers J N . Can first principles calculations aid in lithium-ion battery design?[J]. Journal of Power Sources, 1995, 54(1): 16-19. |
| 36 | Aydinol M K , Kohan A F , Ceder G . Ab initio calculation of the intercalation voltage of lithium-transition-metal oxide electrodes for rechargeable batteries[J]. Journal of Power Sources, 1997, 68(2): 664-668. |
| 37 | Lu H L , Sun S R . Polyimide electrode materials for Li-ion batteries via dispersion-corrected density functional theory[J]. Computational Materials Science, 2018, 146: 119-125. |
| 38 | Aydinol M K , Kohan A F , Ceder G , et al . Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides[J]. Physical Review B Condensed Matter, 1997, 56(3): 1353-1365. |
| 39 | Shi S , Ouyang C Y , Lei M , et al . Effect of Mg-doping on the structural and electronic properties of LiCoO2: a first-principles investigation[J]. Journal of Power Sources, 2007, 171(2): 908-912. |
| 40 | Najafi M . Application of C60, C72 and carbon nanotubes as anode for lithium-ion batteries: a DFT study[J]. Materials Chemistry and Physics, 2017, 195: 195-198. |
| 41 | Hao S , Zhao N , Shi C , et al . Enhanced electrochemical properties of LiCo0.5Ni0.5O2, by Ti-doping: a first-principle study[J]. Ceramics International, 2015, 41(2): 2294-2300. |
| 42 | 宋怀河, 杨树斌, 陈晓红 . 影响锂离子电池高倍率充放电性能的因素[J]. 电源技术, 2009, 33(6): 443-448. |
| Song H H , Yang S B , Chen X H . Factors affecting high rate charge and discharge performance of lithium ion batteries[J]. Chinese Journal of Power Sources, 2009, 33(6): 443-448. | |
| 43 | Shi S , Liu L , Ouyang C Y , et al . Enhancement of electronic conductivity of LiFePO4 by Cr doping and its identification by first-principles calculations[J]. Phys. Rev. B, 2003, 68: 195108. |
| 44 | Ouyang C Y , Shi S Q , Wang Z X , et al . First-principles study of Li ion diffusion in LiFePO4 [J]. Phys. Rev. B, 2004, 69: 104303. |
| 45 | Ouyang C Y , Shi S Q , Wang Z X , et al . The effect of Cr doping on Li ion diffusion in LiFePO4 from first principles investigations and Monte Carlo simulations[J]. Journal of Physics-Condensed Matter, 2004, 16(13): 2265. |
| 46 | 王兆翔, 陈立泉, 黄学杰 . 锂离子电池正极材料的结构设计与改性[J]. 化学进展, 2011, 23(2/3): 284-301. |
| Wang Z X , Chen L Q , Huang X J . First principle calculations of cathode in Li-ion batteries[J]. Progress in Chemistry, 2011, 23(2/3): 284-301. | |
| 47 | Ouyang C Y , Wang D Y , Shi S Q , et al . First principles study on Na x Li1− x FePO4 as cathode material for rechargeable lithium batteries[J]. Chinese Physics Letters, 2006, 23(1): 61-64. |
| 48 | 欧阳楚英 . 锂离子电池正极材料离子动力学性能研究[D]. 北京: 中国科学院物理研究所, 2005. |
| Ouyang C Y . Study on ion dynamics of cathode materials for lithium ion batteries[D]. Beijing: Institute of Physics Chinese Academy of Sciences, 2005. | |
| 49 | Gao L , Xu Z , Zhang S . The co-doping effects of Zr and Co on structure and electrochemical properties of LiFePO4, cathode materials[J]. Journal of Alloys & Compounds, 2017, 739: 529-535. |
| 50 | Yang M Y , Kim S , Kim K , et al . Role of ordered Ni atoms in Li layers for Li-rich layered cathode materials[J]. Advanced Functional Materials, 2017, 27(35): 1700982. |
| 51 | 高玉梅, 杨文鑫, 刘萍 . LiNi0.85- x Co x Mn0.15O2电化学性能的第一性原理研究[J]. 华南师范大学学报(自然科学版), 2017, 49(3): 7-10. |
| Gao Y M , Yang W X , Liu P . First-principles investigation on electrochemical performance of LiNi0.8- x Co x Mn0.15O2 [J]. Journal of South China Normal University (Natural Science Edition), 2017, 49(3): 7-10. | |
| 52 | Dixit M , Markovsky B , Aurbach D , et al . Unraveling the effects of Al doping on the electrochemical properties of LiNi0.5Co0.2Mn0.3O2 using first principles[J]. Journal of the Electrochemical Society, 2017, 164(1): A6359-A6365. |
| 53 | Bianchini F , Fjellvåg H , Vajeeston P . A first-principle investigation of the Li diffusion mechanism in the super-ionic conductor lithium orthothioborate Li3BS3 structure[J]. Materials Letters, 2018, 219: 186-189. |
| 54 | Urquiza M L , Otero M , Luque G L , et al . First-principles studies of silicon underpotential deposition on defective graphene and its relevance for lithium-ion battery materials[J]. Electrochimica Acta, 2016, 208: 92-101. |
| 55 | Rahaman O , Mortazavi B , Rabczuk T . A first-principles study on the effect of oxygen content on the structural and electronic properties of silicon suboxide as anode material for lithium ion batteries[J]. Journal of Power Sources, 2016, 307: 657-664. |
| 56 | Liao N , Zheng B , Zhou H , et al . Effect of carbon segregation on performance of inhomogeneous SiC y O6/5 as anode materials for lithium-ion battery: a first-principles study[J]. Journal of Power Sources, 2016, 334: 39-43. |
| 57 | Luo D , Hou X , Yang J , et al . First principles studies on the electronics structures of (Li0.75Na0.25)(Fe0.75Mn0.25)PO4 cathode materials[J]. Rare Metal Materials & Engineering, 2012, 41(8): 1323-1326. |
| 58 | Chen H , Dawson J , Harding J . Effects of cationic substitution on structural defects in layered cathode materials LiNiO2 [J]. Journal of Materials Chemistry A, 2014, 2(21): 7988-7996. |
| 59 | Huang Z F , Zhang H Z , Wang C Z , et al . First-principles investigation on extraction of lithium ion from monoclinic LiMnO2 [J]. Solid State Sciences, 2010, 40(15): 271-274. |
| 60 | Hoang K . First-principles theory of doping in layered oxide electrode materials[J]. Phys. Rev. Materials, 2017, 1(7): 075403. |
| 61 | Kim S , Hegde V I , Yao Z , et al . First-principles study of lithium cobalt spinel oxides: correlating structure and electrochemistry[J]. ACS Appl. Mater. Interfaces, 2018, 10(16): 13479-13490. |
| 62 | Vallverdu G , Minvielle M , Andreu N , et al . First principle study of the surface reactivity of layered lithium oxides LiMO2 (M = Ni, Mn, Co)[J]. Surface Science, 2016, 649: 46-55. |
| 63 | Min K , Seo S W , Song Y Y , et al . A first-principles study of the preventive effects of Al and Mg doping on the degradation in LiNi0.8Co0.1Mn0.1O2 cathode materials[J]. Physical Chemistry Chemical Physics, 2016, 19(3): 1762. |
| 64 | 王伟东, 仇卫华, 丁倩倩 . 锂离子电池三元材料[M]. 北京: 化学工业出版社, 2015. |
| Wang W D , Qiu W H , Ding Q Q . Lithium-ion Battery Ternary Materials[M]. Beijing: Chemical Industry Press, 2015. | |
| 65 | 夏君磊, 赵世玺 . 锂离子电池电极材料的设计方法及应用[J]. 材料导报, 2001, 15(9): 33-35. |
| Xia J L , Zhao S X . Design method and application of passive electrode material for Li-ion battery[J]. Material Review, 2001, 15(9): 33-35. | |
| 66 | Ullah S , Denis P A , Sato F . Beryllium doped graphene as an efficient anode material for lithium-ion batteries with significantly huge capacity: a DFT study[J]. Applied Materials Today, 2017, 9: 333-340. |
| 67 | Momeni M J , Mousavi-Khoshdel M , Targholi E . First-principles investigation of adsorption and diffusion of Li on doped silicenes: prospective materials for lithium-ion batteries[J]. Materials Chemistry & Physics, 2017, 192: 125-130. |
| 68 | Cui Y , Zhao Y , Chen H , et al . First-principles study of MoO3/graphene composite as cathode material for high-performance lithium-ion batteries[J]. Applied Surface Science, 2018, 433: 1083-1093. |
| 69 | Chen H , Zhang W , Tang X Q , et al . First principles study of P-doped borophene as anode materials for lithium ion batteries[J]. Applied Surface Science, 2018, 427: 198-205. |
| 70 | Wang H , Wu M , Lei X , et al . Siligraphene as a promising anode material for lithium-ion batteries predicted from first-principles calculations[J]. Nano Energy, 2018, 49: 67-76. |
| 71 | Jiang H R , Zhao T S , Liu M , et al . Two-dimensional SiS as a potential anode material for lithium-based batteries: a first-principles study[J]. Journal of Power Sources, 2016, 331: 391-399. |
| 72 | Cho E , Seo S W , Min K . Theoretical prediction of surface stability and morphology of LiNiO2 cathode for Li ion battery[J]. ACS Applied Materials & Interfaces, 2017, 9(38): 33257-33266. |
| 73 | Fan L , Zhuang H L , Gao L , et al . Regulating Li deposition at artificial solid electrolyte interphases[J]. Journal of Materials Chemistry A, 2017, 5(7): 3483-3492. |
| 74 | Vajeeston P . Ionic conductivity enhancement by particle size reduction in Li2FeSiO4 [J]. Materials Letters, 2018, 218: 313-316. |
| 75 | 龚鑫 . 第一性原理计算研究锂离子电池正极材料LiCoO2的热力学性能[D]. 南昌: 江西师范大学, 2014. |
| Gong X . First-principles calculation study of the thermodynamic properties of LiCoO2 cathode material for lithium ion batteries[D]. Nanchang: Jiangxi Normal University, 2014. | |
| 76 | 刘兆君 . 锂离子电池电极材料中热力学和动力学问题的第一性原理研究[D]. 北京: 中国科学院物理研究所, 2010. |
| Liu Z J . First-principles study of thermodynamic and kinetic problems in lithium ion battery electrode materials[D]. Beijing: Institute of Physics Chinese Academy of Sciences, 2010. | |
| 77 | Ali S , Rashid M , Hassan M , et al . Ab-initio study of electronic, magnetic and thermoelectric behaviors of LiV2O4, and LiCr2O4, using modified Becke-Johson (mBJ) potential[J]. Physica B Condensed Matter, 2018, 537: 329-335. |
| [1] | Yuanchao LIU, Bin GUAN, Jianbin ZHONG, Yifan XU, Xuhao JIANG, Duan LI. Investigation of thermoelectric transport properties of single-layer XSe2 (X=Zr/Hf) [J]. CIESC Journal, 2023, 74(9): 3968-3978. |
| [2] | Jing LI, Conghao SHEN, Daliang GUO, Jing LI, Lizheng SHA, Xin TONG. Research progress in the application of lignin-based carbon fiber composite materials in energy storage components [J]. CIESC Journal, 2023, 74(6): 2322-2334. |
| [3] | Zhongliang XIAO, Bilu YIN, Liubin SONG, Yinjie KUANG, Tingting ZHAO, Cheng LIU, Rongyao YUAN. Research progress of waste lithium-ion battery recycling process and its safety risk analysis [J]. CIESC Journal, 2023, 74(4): 1446-1456. |
| [4] | Feng DU, Siqi YIN, Hui LUO, Wenan DENG, Chuan LI, Zhenwei HUANG, Wenjing WANG. Study on size effect of H2 adsorption and dissociation on Mo x S y clusters [J]. CIESC Journal, 2022, 73(9): 3895-3903. |
| [5] | Xiaqi YU, Ge FENG, Jinyan ZHAO, Jiayuan LI, Shengwei DENG, Jingnan ZHENG, Wenwen LI, Yaqiu WANG, Lan SHEN, Xu LIU, Weiwei XU, Jianguo WANG, Shibin WANG, Zihao YAO, Chengli MAO. A first-principles study of the interaction between TDI-TMP-T313 and AP [J]. CIESC Journal, 2022, 73(8): 3511-3517. |
| [6] | Jihao ZHAO, Weiqiang TANG, Xiaofei XU, Shuangliang ZHAO, Jionghao HE. Adsorption energy of bonding agent on nano-filler in polymer composites [J]. CIESC Journal, 2022, 73(7): 3174-3181. |
| [7] | Xiaokun HE, Yuan XUE, Ran ZUO. Quantum chemistry study on gas reaction path in InN MOCVD growth [J]. CIESC Journal, 2022, 73(12): 5638-5647. |
| [8] | Pengpeng WANG, Yanggang JIA, Xia SHAO, Jie CHENG, Aiqin MAO, Jie TAN, Daolai FANG. Preparation and lithium storage performance of K+-doped spinel (Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4 high-entropy oxide anode materials [J]. CIESC Journal, 2022, 73(12): 5625-5637. |
| [9] | Xiaosong LUO, Jinbao HUANG, Mei ZHOU, Xin MU, Weiwei XU, Lei WU. Theoretical study on the mechanism of hydrolysis/alcoholysis/ammonolysis of butanediol terephthalate dimer [J]. CIESC Journal, 2022, 73(11): 4859-4871. |
| [10] | Liubin SONG, Yixuan WANG, Yinjie KUANG, Yubo XIA, Zhongliang XIAO. Development and prospect of pivotal materials and technologies in sodium-ion batteries [J]. CIESC Journal, 2022, 73(11): 4814-4825. |
| [11] | Xiang GONG, Linsen LI, Zhao JIANG. Employing PdCo/SiO2 catalyst in high activity dehydrogenation reaction of heterocyclic H2 storage carrier [J]. CIESC Journal, 2022, 73(10): 4448-4460. |
| [12] | Xianhui ZHU, Fu WANG, Jiecheng XIA, Jinliang YUAN. Density functional theory investigation on the NH3 and CO2 absorption by functional ionic liquids [J]. CIESC Journal, 2022, 73(10): 4324-4334. |
| [13] | Yiwei ZHOU, Zhuo CHEN, Jianhong XU. Progress and prospect of recycling spent lithium battery cathode materials by hydrometallurgy [J]. CIESC Journal, 2022, 73(1): 85-96. |
| [14] | Shenggui MA, Bowen TIAN, Yuwei ZHOU, Lin CHEN, Xia JIANG, Tao GAO. DFT study of adsorption of H2S on N-doped Stone-Wales defected graphene [J]. CIESC Journal, 2021, 72(9): 4496-4503. |
| [15] | ZHANG Fangfang, HAN Min, ZHAO Juan, LING Lixia, ZHANG Riguang, WANG Baojun. DFT study on reduction of NO over Pd atom anchored on single-vacancy graphene [J]. CIESC Journal, 2021, 72(3): 1382-1391. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||

