| 1 |
Han J , Wang X , Yue J , et al . Catalytic upgrading of coal pyrolysis tar over char-based catalysts[J]. Fuel Processing Technology, 2014, 122: 98-106.
|
| 2 |
Rombi E , Cutrufello M G , Atzori L , et al . CO methanation on Ni-Ce mixed oxides prepared by hard template method[J]. Applied Catalysis A: General, 2016, 515: 144-153.
|
| 3 |
Wang S G , Cao D B , Li Y W , et al . CO2 reforming of CH4 on Ni (111): a density functional theory calculation[J]. The Journal of Physical Chemistry B, 2006, 110(20): 9976-9983.
|
| 4 |
Wang S G , Liao X Y , Hu J , et al . Kinetic aspect of CO2 reforming of CH4 on Ni(111): a density functional theory calculation[J]. Surface Science, 2007, 601(5): 1271-1284.
|
| 5 |
Solomon P R , Serio M A , Carangelo R M , et al . Analysis of the Argonne premium coal samples by thermogravimetric Fourier transform infrared spectroscopy[J]. Energy & Fuels, 1990, 4(3): 319-333.
|
| 6 |
Choe S J , Kang H J , Park D H , et al . Adsorption and dissociation reaction of carbon dioxide on Ni (1 1 1) surface: molecular orbital study[J]. Applied Surface Science, 2001, 181(3/4): 265-276.
|
| 7 |
Kresse G , Furthmüller J . Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169.
|
| 8 |
Kresse G , Furthmüller J . Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1): 15-50.
|
| 9 |
Blöchl P E . Projector augmented-wave method[J]. Physical Review B, 1994, 50(24): 17953-17979.
|
| 10 |
Perdew J P , Burke K , Ernzerhof M . Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865.
|
| 11 |
Kresse G , Joubert D . From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59(3): 1758.
|
| 12 |
Sheppard D , Xiao P , Chemelewski W , et al . A generalized solid-state nudged elastic band method[J]. J. Chem. Phys., 2012, 136(7): 074103.
|
| 13 |
Kong L , Li G , Jin L , et al . Pyrolysis behaviors of two coal-related model compounds on a fixed-bed reactor[J]. Fuel Processing Technology, 2015, 129: 113-119.
|
| 14 |
Li L , Fan H , Hu H . A theoretical study on bond dissociation enthalpies of coal based model compounds[J]. Fuel, 2015, 153: 70-77.
|
| 15 |
Wang M F , Zuo Z J , Ren R P , et al . Theoretical study on catalytic pyrolysis of benzoic acid as a coal-based model compound[J]. Energy & Fuels, 2016, 30(4): 2833-2840.
|
| 16 |
凌丽霞, 赵俐娟, 章日光, 等 . 苯甲酸和苯甲醛热解机理的量子化学研究[J]. 化工学报, 2009, 60(5): 1224-1230.
|
|
Ling L X , Zhao L J , Zhang R G , et al . Pyrolysis mechanisms of benzoic acid and benzaldehyde based on quantum chemistry[J]. CIESC Journal, 2009, 60(5): 1224-1230.
|
| 17 |
Xu B , Lu W , Sun Z , et al . High-quality oil and gas from pyrolysis of Powder River Basin coal catalyzed by an environmentally-friendly, inexpensive composite iron-sodium catalysts[J]. Fuel Processing Technology, 2017, 167: 334-344.
|
| 18 |
Rodriguez J A , Hanson J C , Frenkel A I , et al . Experimental and theoretical studies on the reaction of H2 with NiO: role of O vacancies and mechanism for oxide reduction[J]. Journal of the American Chemical Society, 2002, 124(2): 346-354.
|
| 19 |
Selcuk S , Selloni A . DFT+U study of the surface structure and stability of Co3O4(110): dependence on U[J]. The Journal of Physical Chemistry C, 2015, 119(18): 9973-9979.
|
| 20 |
Wang L , Maxisch T , Ceder G . Oxidation energies of transition metal oxides with in the GGA+U framework[J]. Physical Review B, 2006, 73(19): 195107.
|
| 21 |
Dudarev S L , Botton G A , Savrasov S Y , et al . Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+ U study[J]. Physical Review B, 1998, 57(3): 1505.
|
| 22 |
Hüfner S . Electronic structure of NiO and related 3d-transition-metal compounds[J]. Advances in Physics, 1994, 43(2): 183-356.
|
| 23 |
Yan M , Chen S P , Mitchell T E , et al . Atomistic studies of energies and structures of (hk0) surfaces in NiO[J]. Philosophical Magazine A, 1995, 72(1): 121-138.
|
| 24 |
Li L , Kanai Y . Antiferromagnetic structures and electronic energy levels at reconstructed NiO(111) surfaces: ADFT+U study[J]. Physical Review B, 2015, 91(23): 235304.
|
| 25 |
Zeng Y , Ma H , Zhang H , et al . Ni-Ce-Al composite oxide catalysts synthesized by solution combustion method: enhanced catalytic activity for CO methanation[J]. Fuel, 2015, 162: 16-22.
|
| 26 |
Kresse G , Hafner J . Ab initio molecular dynamics for liquid metals[J]. Physical Review B, 1993, 47(1): 558-561.
|
| 27 |
Rohrbach A , Hafner J , Kresse G . Molecular adsorption on the surface of strongly correlated transition-metal oxides: a case study for CO/NiO(100)[J]. Physical Review B, 2004, 69(7): 075413.
|
| 28 |
Wang B , Nisar J , Ahuja R . Molecular simulation for gas adsorption at NiO (100) surface[J]. ACS Applied Materials & Interfaces, 2012, 4(10): 5691-5697.
|
| 29 |
Eskay T P , Britt P F , Buchanan III A C . Pyrolysis of coal model compounds containing aromatic carboxylic acids: the role of carboxylic acids in cross-linking reactions in low-rank coal[R]. Oak Ridge National Lab., TN (United States), 1997.
|
| 30 |
Eskay T P , Britt P F , Buchanan A C . Does decarboxylation lead to cross-linking in low-rank coals?[J]. Energy & Fuels, 1996, 10(6): 1257-1261.
|
| 31 |
Manion J A , McMillen D F , Malhotra R . Decarboxylation and coupling reactions of aromatic acids under coal-liquefaction conditions[J]. Energy & Fuels, 1996, 10(3): 776-788.
|
 ),刘江涛,赵月,黄伟,左志军(
),刘江涛,赵月,黄伟,左志军( 京公网安备 11010102001995号
京公网安备 11010102001995号


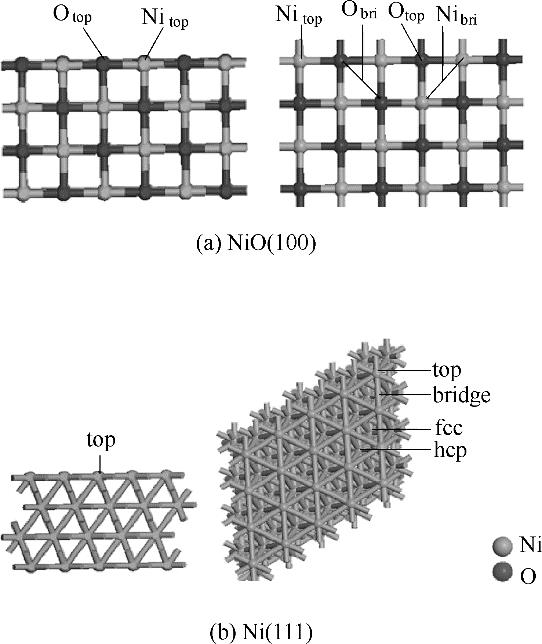
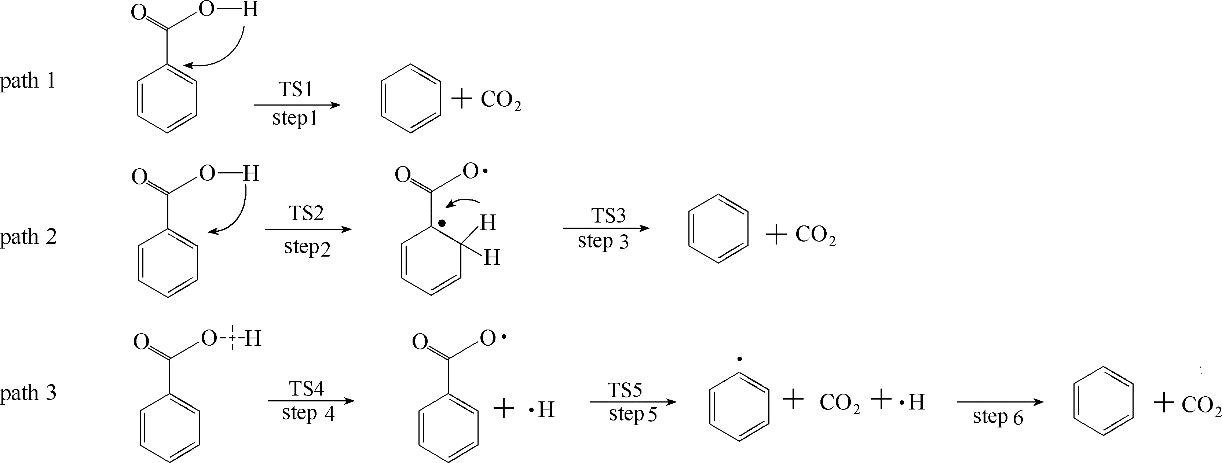
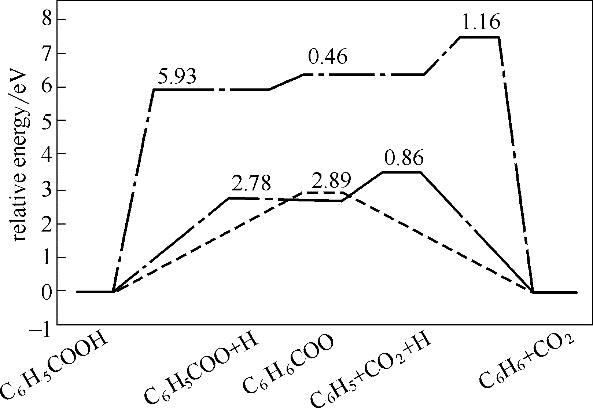


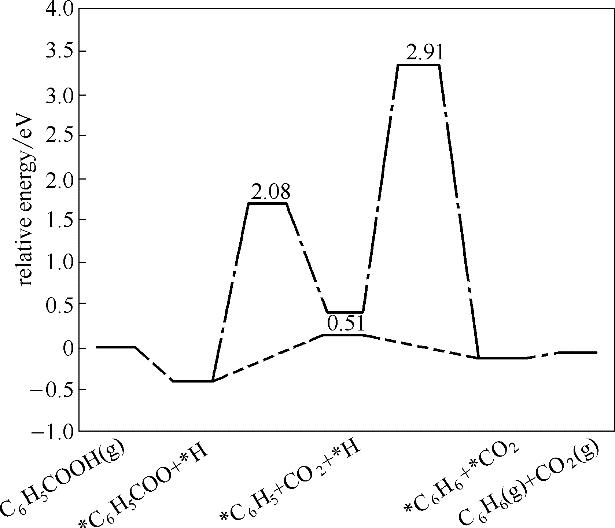



 分析
分析