| 1 |
Lei Z , Yang D , Zhang Y H , et al . Constructions of coal and char molecular models based on the molecular simulation technology[J]. Journal of Fuel Chemistry & Technology, 2017, 45(7): 769-779.
|
| 2 |
Xie K C . Structure and Reactivity of Coal [M]. New York: Springer-Verlag Press, 2015: 1-27.
|
| 3 |
Mathews J P , Chaffee A L . The molecular representations of coal— a review[J]. Fuel, 2012, 96(7): 1-14.
|
| 4 |
Carlson G A . Computer simulation of the molecular structure of bituminous coal[J]. Energy & Fuels, 1992, 6(6): 771-778.
|
| 5 |
Zhang Z , Kang Q , Wei S , et al . Large scale molecular model construction of Xishan bituminous coal[J]. Energy & Fuels, 2017, 31: 1310-1317.
|
| 6 |
Mathews J P , Sharma A . The structural alignment of coal and the analogous case of Argonne Upper Freeport coal[J]. Fuel, 2012, 95(1): 19-24.
|
| 7 |
相建华, 曾凡桂, 李彬, 等 . 成庄无烟煤大分子结构模型及其分子模拟[J]. 燃料化学学报, 2013, 41(4): 391-399.
|
|
Xiang J H, Zeng F G, Li B, et al Construction of macromolecular structural model of anthracite from Chengzhuang coal mine and its molecular simulation[J]. Journal of Chemistry and Technology, 2013, 41(4): 391-399.
|
| 8 |
Behar F , Hatcher P G . Artificial coalification of a fossil wood from brown coal by confined system pyrolysis[J]. Fuel & Energy Abstracts, 1996, 37(3): 984-994.
|
| 9 |
Salmon E , van Duin Act, Lorant F , et al . Early maturation processes in coal (2): Reactive dynamics simulations using the ReaxFF reactive force field on Morwell Brown coal structures[J]. Organic Geochemistry, 2009, 40(12): 1195-1209.
|
| 10 |
Heek K H V , Hodek W . Structure and pyrolysis behaviour of different coals and relevant model substances[J]. Fuel, 1994, 73(6): 886-896.
|
| 11 |
Mushtaq F , Mat R , Ani F N . A review on microwave assisted pyrolysis of coal and biomass for fuel production[J]. Renewable & Sustainable Energy Reviews, 2014, 39(6): 555-574.
|
| 12 |
Abdelsayed V , Shekhawat D , Smith M W , et al . Microwave-assisted pyrolysis of Mississippi coal: a comparative study with conventional pyrolysis[J]. Fuel, 2018, 217: 656-667.
|
| 13 |
Gao S , Zhai L , Qin Y , et al . Investigation into the cleavage of chemical bonds induced by CO2 and its mechanism during the pressurized pyrolysis of coal[J]. Energy & Fuels, 2018, 32(3): 3243-3253.
|
| 14 |
陈兆辉, 高士秋, 许光文 . 煤热解过程分析与工艺调控方法[J]. 化工学报, 2017, 68(10): 3693-3707.
|
|
Chen Z H , Gao S Q , Xu G W . Analysis and control methods of coal pyrolysis process[J]. CIESC Journal, 2017, 68(10): 3693-3707.
|
| 15 |
van Duin Act, Dasgupta S , Lorant F , et al . ReaxFF: a reactive force field for hydrocarbons[J]. Journal of Physical Chemistry A, 2001, 105(41): 9396-9409.
|
| 16 |
Salmon E , van Duin Act, Lorant F , et al . Thermal decomposition process in algaenan of Botryococcus braunii race L. (2): Molecular dynamics simulations using the ReaxFF reactive force field[J]. Organic Geochemistry, 2009, 40(3): 416-427.
|
| 17 |
Wang H , Feng Y , Zhang X , et al . Study of coal hydropyrolysis and desulfurization by ReaxFF molecular dynamics simulation[J]. Fuel, 2015, 145: 241-248.
|
| 18 |
Wang J P , Li G Y , Guo R , et al . Theoretical and experimental insight into coal structure: establishing a chemical model for Yuzhou lignite[J]. Energy & Fuels, 2016, 31(1): 124-132.
|
| 19 |
Zhan J H , Wu R , Liu X , et al . Preliminary understanding of initial reaction process for subbituminous coal pyrolysis with molecular dynamics simulation[J]. Fuel, 2014, 134(9): 283-292.
|
| 20 |
Hong D , Guo X . Molecular dynamics simulations of Zhundong coal pyrolysis using reactive force field[J]. Fuel, 2017, 210: 58-66.
|
| 21 |
Li W , Zhu Y , Chen S , et al . Research on the structural characteristics of vitrinite in different coal ranks[J]. Fuel, 2013, 107(9): 647-652.
|
| 22 |
朱学栋, 朱子彬, 韩崇家, 等 . 煤中含氧官能团的红外光谱定量分析[J]. 燃料化学学报, 1999, (4): 335-339.
|
|
Zhu X D , Zhu Z B , Han C J , et al . Quantitative determination of oxygen containing functional groups in coal by FTIR spectroscopy[J]. Journal of Fuel Chemistry and Technology, 1999, (4): 335-339.
|
| 23 |
Hayashi J , Takahashi H , Doi S , et al . Reactions in brown coal pyrolysis responsible for heating rate effect on tar yield[J]. Energy & Fuels, 2000, 14(2): 400-408.
|
| 24 |
Yan G C , Zhang Z Q , Yan K F . Reactive molecular dynamics simulations of the initial stage of brown coal oxidation at high temperatures[J]. Molecular Physics, 2012, 111(1): 147-156.
|
| 25 |
Chen B , Diao Z J , Zhao Y L , et al . A ReaxFF molecular dynamics (MD) simulation for the hydrogenation reaction with coal related model compounds[J]. Fuel, 2015, 154(5): 114-122.
|
| 26 |
Bhoi S , Banerjee T , Mohanty K . Molecular dynamic simulation of spontaneous combustion and pyrolysis of brown coal using ReaxFF[J]. Fuel, 2014, 136(6): 326-333.
|
| 27 |
Zhou Z , Guo L , Chen L , et al . Study of pyrolysis of brown coal and gasification of coal-water slurry using the ReaxFF reactive force field[J]. International Journal of Energy Research, 2018, 42(7): 2465-2480.
|
| 28 |
刘振宇 . 煤化学的前沿与挑战: 结构与反应[J]. 中国科学: 化学, 2014, 44(9): 1431-1438.
|
|
Liu Z Y . Advancement in coal chemistry: structure and reactivity[J]. Scientia Sinica Chimica, 2014, 44(9): 1431-1438.
|
| 29 |
张小盟, 李宁煜, 高文青 . 宁夏煤炭利用效率的影响因素分析[J]. 煤炭加工与综合利用, 2017, (3): 59-65.
|
|
Zhang X M , Li N Y , Gao W Q . Analysis on the factors affecting the utilization efficiency of coal in Ningxia[J]. Coal Processing and Comprehensive Utilization, 2017, (3): 59-65.
|
| 30 |
李壮楣, 王艳美, 李平, 等 . 宁东红石湾煤大分子模型构建及量子化学计算[J]. 化工学报, 2018, 69(5): 2208-2216.
|
|
Li Z M , Wang Y M , Li P , et al . Macromolecular model construction and quantum chemical calculation of Ningdong Hongshiwan coal[J]. CIESC Journal, 2018, 69(5): 2208-2216.
|
| 31 |
Zhao Y , Truhlar D G . The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals[J]. Theor. Chem. Acc., 2008, 120: 215-241.
|
| 32 |
Weismiller M R , van Duin Act, Lee J , et al . ReaxFF reactive force field development and applications for molecular dynamics simulations of ammonia borane dehydrogenation and combustion[J]. Journal of Physical Chemistry A, 2010, 114: 5485-5492.
|
| 33 |
Zheng M , Li X , Liu J , et al . Pyrolysis of Liulin coal simulated by GPU-based ReaxFF MD with cheminformatics analysis[J]. Energy & Fuels, 2014, 28(1): 522–534.
|
| 34 |
张莉 . 五牧场11号煤结构模型构建及其超分子特征[D]. 太原: 太原理工大学, 2013.
|
|
Zhang L . Molecular structure model building and supermolecule characteristic of Wumuchang No.11 coal[D]. Taiyuan: Taiyuan University of Technology, 2013.
|
| 35 |
马伦, 陆大荣, 梁汉东, 等 . 神华长焰煤大分子结构特征的研究[J]. 燃料化学学报, 2013, 41(5): 513-522.
|
|
Ma L , Lu D R , Liang H D , et al . Preliminary study on macromolecular structure characteristics of Shenhua long flame coal[J]. Journal of Fuel Chemistry & Technology, 2013, 41(5): 513-522.
|
| 36 |
Zheng M , Li X , Nie F , et al . Investigation of overall pyrolysis stages for Liulin bituminous coal by large-scale ReaxFF molecular dynamics[J]. Energy & Fuels, 2017, 31(4): 3675−3683.
|
| 37 |
Li W , Zhu Y M , Wang G , et al . Molecular model and ReaxFF molecular dynamics simulation of coal vitrinite pyrolysis[J]. Journal of Molecular Modeling, 2015, 21(8): 188-200.
|
 ),高红凤,王贵,吴浪浪,许靖钦,李壮楣,李平,白红存(
),高红凤,王贵,吴浪浪,许靖钦,李壮楣,李平,白红存( 京公网安备 11010102001995号
京公网安备 11010102001995号



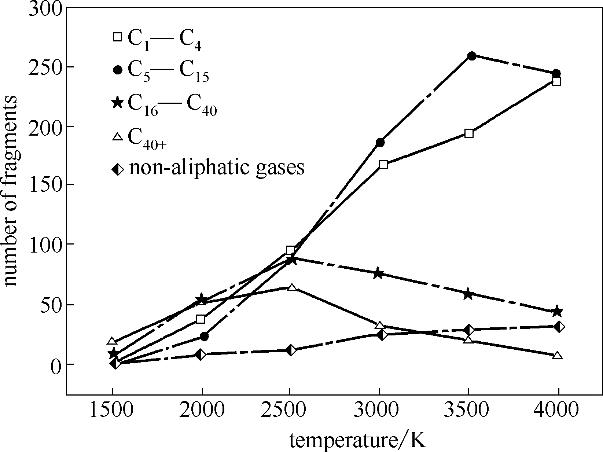
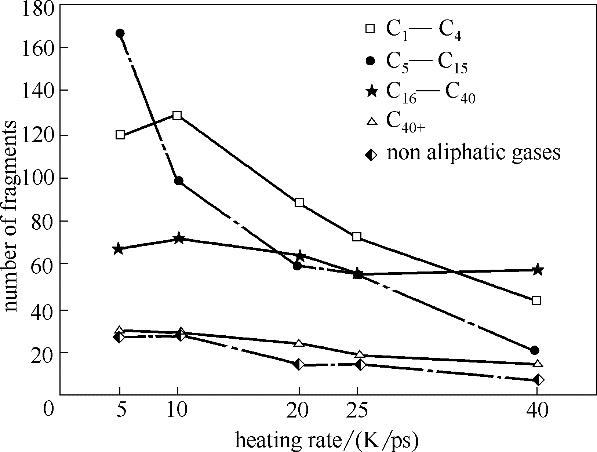
 分析
分析